|
Упрощенный метод получения кДНК низкокопийных и молчащих эукариотических генов на примере реналазы человека
Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, Ключевые слова: : кДНК; ПЦР; экзон; клонирование; экспрессия DOI: 10.18097/BMCRM00101 ВВЕДЕНИЕ Классические методы клонирования кДНК нового гена требуют выделения и очистки поли-А селективной мРНК из клеток или тканей с экспрессирующимся геном и последующим синтезом кДНК [1-3]. Выделение низкокопийных мРНК связано с определенными трудностями, а в случае тканей человека сопряжено с соблюдением юридически установленных процедур. Клонирование молчащих генов таким способом и вовсе невозможно. В настоящее время анализ геномных последовательностей млекопитающих по определенным алгоритмам (gene prediction method) позволяют с достаточной точностью характеризовать геномные структуры. Наличие банка данных геномных последовательностей ДНК различных животных и человека (https://www.ncbi.nlm.nih.gov/nucleotide/), а также использование достаточно рутинной процедуры получениz ПЦР-продуктов любых участков из геномной ДНК и последующем объединением таких участков ДНК позволяет достаточно быстро конструировать любые генетические конструкции с известной нуклеотидной последовательностью. Методы ПЦР широко используются при клонировании различных нуклеотидных последовательностей и создания гибридных генов [4-6]. Для объединения двух фрагментов ДНК методом ПЦР в начале получают первый ПЦР-фрагмент, у которого с помощью синтетического олигонуклеотидного праймера на 5’-конце достраивают участок ДНК (около 20 нуклеотидов), комплементарный второму предыдущего 3’-концевому фрагменту ДНК. В результате отжига первого экзонного фрагмента со вторым экзоновым фрагментом осуществляется амплификация объединяемого фрагмента ДНК. В 1992 г. Booth с соавторами [7] амплифицировали индивидуальные экзоны человеческого цилиарного нейротрофического фактора (human ciliary neurotrophic factor - CNTF), содержащего всего два экзона и один интрон. Были получены ПЦР-продукты первого и второго экзона, а затем эти экзоны объединяли и встраивали в плазмидный вектор с получением полной кодирующей последовательности CNTF. В этом случае была использована технология так называемого безлигированного клонирования (ligation-independent cloning - без использования эндонуклеаз рестрикций и лигазы) [8, 9]. Такая технология основывается на использовании олигонуклеотидных праймеров, в которых на 5’-конце дезокситимидиновые остатки заменяют дезоксиуроциловыми остатками. После ПЦР ампликоны обрабатывают урацил-ДНК-гликозилазой (uracil DNA glycosylase – UDG), которая генерирует в ампликонах 3’-выступающие «липкие» (U)n -концы. По этим концам происходит нековалентное объединение ПЦР-фрагментов и безковалентное встраивание их в экспрессионный вектор. Однако использование такой технологии объединения трех и более экзонов для получения полной кодирующей последовательности гена в современной литературе не описано. В 2017 г. An с соавторами [10] описали метод получения полной кодирующей последовательности (кДНК) гена PIGR (human polymeric immunoglobulin receptorv) человека, включающего 10 экзонов, посредством объединения единичных экзонов, полученных с матрицы ДНК при помощи ПЦР метода (Genomic DNA Splicing). Вначале авторы амплифицировали индивидуальные экзоны с матрицы геномной ДНК в раздельных реакционных средах с применением оптимизированных прямого и обратного праймеров в области интрона, которые фланкировали синтезируемые экзоны. После проведения амплификации экзонов ПЦР ампликоны подвергали электрофоретической очистке в агарозном геле. Во втором раунде ПЦР амплификации получали отдельные экзоны без интронных ДНК последовательностей. Для этого использовали праймеры, содержащие нуклеотидные последовательности, перекрывающие соседние экзоны. Затем проводили объединение экзонов ПЦР методом, получая полноразмерную последовательность кДНК. Особенность такого подхода заключается в том, что для получения отдельных экзонов использовали двухэтапную амплификацию и, следовательно, удвоенное количество праймеров для синтеза каждого экзона и с обязательной очисткой ДНК ампликонов. В этом же году (2017) Davies с соавторами [11] так же применили «SPLICE» метод при получении полноразмерной кДНК последовательности гена SWS1 (short-wavelength-sensitive 1), содержащего 5 экзонов, из геномной ДНК лемура. Сначала в ходе ПЦР амплификации авторы получали индивидуальные экзоны без интронных последовательностей с матрицы геномной ДНК. Прямые и обратные праймеры были подобраны таким образом, чтобы у ампликонов в начале второго экзона находился конец первого экзона. В первом раунде амплификации получали отдельные экзоны, а во втором раунде получали парные экзоны 1-2, 2-3, 3-4, 4-5. Такие ампликоны очищали электрофоретически в агарозном геле, а затем проводили их клонирование с последующим секвенированием. В третьем раунде получали ПЦР-продукты 1-3 и 3-5 экзонов. И на заключительной стадии – ПЦР-продукт полной последовательности кДНК (1-5 экзоны). При таком подходе при получении кДНК использовалась трудоемкая работа по очистки каждого ПЦР-продукта, его клонирование с последующим секвенированием. Развитие химически-автоматического олигонуклеотидного синтеза позволило удлинять олигонуклеотидную матрицу при помощи ПЦР до несколько сотен нуклеотидов. Стратегия ступенчатого наращивания нуклеотидной последовательности в результате нескольких ПЦР позволила осуществить синтез гена длиной 400 пар оснований (п.о., b.p.) [12]. В работе Гуревич и др. удалось осуществить химический синтез гена на синтетической матрице длиной более 650 п.о. [13]. Однако химический синтез гена на данный момент является трудоемкой и дорогостоящей работой и для получения полных кодирующих областей генов широко не используется. Для получения кДНК гена эукариот мы предлагаем упрощенный экзоновый метод получения кодирующей последовательности гена непосредственно с геномной ДНК посредством синтеза отдельных экзонов с последующим их объединением. Такой подход можно использовать для эффективного и быстрого клонирования функционально молчащих генов и генов с низким уровнем экспрессии. Метод основан на получении рекомбинантного гена из геномной ДНК организма по его известным экзонным структурам, что позволяет собрать любой ген из банка данных библиотеки генов эукариот. Суть его состоит в том, что вначале синтезируются ПЦР продукты отдельных экзонов, которые включают перекрывающие участки смежных экзонов. На следующих этапах амплификации идет объединение экзонов без предварительной очистки ПЦР продуктов от предыдущий стадии амплификации экзонов. Такая технология упрощенного «экзонового» метода была применена нами при клонировании гена реналазы человека. МАТЕРИАЛЫ И МЕТОДЫ
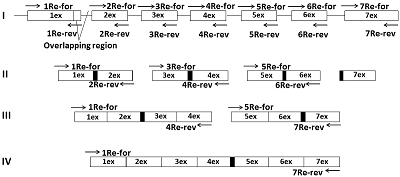
Реактивы Для исследования были использованы следующие реагенты: Tersus-ДНК полимераза («Евроген», Россия), эдонуклеазы рестрикции BamHI, XhoI и маркеры молекулярной массы («Ферментас», Латвия), Ni-сефароза («GE Healtheare», Швеция). Система для очистки фрагментов ДНК (Wizard® SV Gel и PCR Clean-Up) и TA-вектор (pGEM®-T easy vector) были преобретены в («Promega», USA). Плазмидный экспрессионный вектор pET-28a(f+) получен от «Novagen» (Англия). Олигонуклеотиды для ПЦР (табл. 1) были синтезированы и очищены через ПААГ («Синтол», Россия). E. coli штаммы Rosetta (DE3) были куплены в «Novagen» (Англия). Другие химические реактивы были приобретены у «Sigma-Aldrich» (Россия). Получение матрицы ДНК человека Геномную ДНК человека получали из лимфоцитов крови человека стандартным фенол-хлороформом методом экстракции [14, 15]. В работе использовали 0.5 мл цельной крови, полученной от здорового донора. Образец цельной крови смешивали с 0.5 мл 20 мМ трис-ацетатного буфера, pH 7.5, содержащего 22% сахарозу, 20 мМ MgCl2, 1% Triton X-100 и центрифугировали 10 мин при 3800 об/мин, используя центрифугу Eppendorf 5415R (Германия). Осадок клеток ресуспендировали в том же буфере, разбавляли в два раза дистиллированной водой и центрифугировали, как указано выше. Полученный клеточный осадок ресуспендировали в 0.9 мл 10 мМ трис-HCl-буфера, рН 7.5, содержащего 400 мМ NaCl и 2 мМ ЭДТА. После добавления 0.1 мл 10% SDS и протеиназы К (конечная концентрация 1 мг/мл) полученную смесь инкубировали при 58°С в течение 3 ч при перемешивании и затем последовательно обрабатывали одним объемом фенола и затем одним объемом фенол-хлороформа (1:1). После добавления 0.1 объема 3 М ацетата натрия, рН 5.0 и 0.6 объема изопропанола ДНК осаждали центрифугированием при 12000об/мин в течение 15 мин с использованием той же центрифуги и затем промывали в холодном 70% этаноле. Очищенную ДНК растворяли в 200 мкл H2O, не содержащей РНКазы, и ее концентрацию ДНК измеряли по поглощению при 260 нм с использованием УФ спектрофотометра Cary 50 (США). Образцы ДНК хранили при -20°С. ПЦР метод ПЦР проводили в 20 мкл реакционной смеси, содержащей 6.7 мМ трис pH 8.8, 1.66 мМ (NH4)2SO4; 0.02% Twin-20; 1 мМ MgCl2; 0.2 мМ dNTP; 1% глицерин, 4 мкМ (прямой и обратный) праймеры, 50 – 100 нг ДНК (или по 1 мкл ампликона от ПЦР) и 0.5 ед. Tersus-ДНК-полимеразы. Условия ПЦР: денатурация – 95ºС в течение 5 мин – 1 цикл; элонгация – 92º С – 20 с, 55ºС – 15 с, 72ºС – от 30 с до 2 – 3 мин; количество циклов от 30 до 35; заключительный цикл 72ºС – 3 мин. Время элонгации с Tersus-ДНК-полимеразой зависит от длины синтезируемого фрагмента. Синтез одиночных экзонов время элонгации – 30 – 40 сек, двойных экзонов – 1 мин, для четырех экзонов – 2 мин, при синтезе полноразмерной кДНК – 4 мин. При реамплификации матрицы ДНК использовали те же условия. Электрофорез фрагментов ДНК проводили в 2% агарозном геле с использованием трис-боратного буфера рН 8.0 [16].Электрофорез в 10% ПААГ проводили по Лэммли [17]. Клонирование кДНК гена реналазы вначале проводили с плазмидой pGEM-T. Этот вектор включает на 3’-концах выступающий единичный Т-остаток, который значительно усиливает лигирование ПЦР продукта. В результате рестриктного анализа с последующим секвенированием отбирался клон, содержащий исходную нуклеотидную последовательность гена реналазы. Данную последовательность вырезали по сайтам рестрикции BamHI/XhoI и переклонировали в плазмиду pET-28a(+). Трансформацию клеток E. coli. проводили по методу Kushner [18]; аликвоты компетентных клеток (по 0.2 мл) хранили при -70ºС. Трансформацию проводили по стандартному методу Cohen [19], используя 1 мкл вектора. Трансформированные клетки высевали на чашки с LB-агаром, содержащий антибиотик необходимый для селекции. Экспрессию клонированного гена проводили следующим образом. Колонию клеток после трансформации, подращивали в 4 мл LB-среды с 50 мкг/мл каномицина (Км) в течение ночи. Ночную культуру пересевали в 200 мл этой же LB среды и наращивали культуру клеток до оптической плотности 0.5 – 0.7 ед. Затем вносили ИПТГ до концентрации 1.0 мМ и продолжали инкубировать клетки при интенсивном перемешивании еще 3 ч. Биомассу собирали центрифугированием при 5000 g 4° С течение 20 мин. на Avanti J-E центрифуге («Beckman Coulter», США) и хранили при -20ºС. Выделение рекомбинантного белка осуществляли методом аффинной хроматографии с использованием Ni-сефарозы в 8 М буферном растворе мочевины. Биомассу, содержащую рекомбинантный белок, денатурировали в растворе, содержащем 8 М мочевину, 0.1 М NaH2PO4, 0.01 М трис, рН 8.0. Лизат переносили в колбу, добавляли к нему суспензию Ni-сефарозы (1 мл суспензии на 25 мл лизата) и инкубировали 2 ч при комнатной температуре в шейкере с легким покачиванием (40 об/мин). Полученную суспензию пропускали через колонку с использованием хроматографической системы Biologic LP («Bio-Rad», Германия). Отмывку сорбента со связавшимся белком осуществляли тем же буфером, который использовали при денатурации биомассы, но с pH 6.3 , а затем с pH 5.9. Элюцию проводили с использованием аналогичного буферного раствора, pH 4.5. Препараты очищенных рекомбинантных белков растворяли в 50 мМ буферном растворе трис-HCl (рН 9.0) с помощью ступенчатого диализа при температуре 4°С против нескольких перемен буферного раствора (50 мМ трис-HCl, рН 9.0), содержащих 6 М, 4 М, 2 М и 1 М мочевину, а затем против буферного раствора, не содержащего мочевину. Концентрацию белка определяли спектрофотометрически. Секвенирование ДНК проводили на капиллярном ДНК секвенаторе (310 DNA analyzer, «Applied Biosystems», США) с использованием BigDye Terminator v 3,1 Cycle Sequencing Kit в соответствии с рекомендациями фирмы производителя. Полученную последовательность ДНК соотносили с геном реналазы (NM_001031709, GeneBank), используя программу BLAST, доступную на сайте National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). РЕЗУЛЬТАТЫ Согласно нуклеотидной последовательности банка данных (код доступа GenBank www.ncbi. Homo sapiens NM_001031709), ген реналазы включает семь кодирующих областей (экзонов). Следовательно, для синтеза полной кодирующей последовательности реналазы методом ПЦР необходимо было синтезировать 14 праймеров (семь «прямых» и семь «обратных»). Для подборки праймеров использовали следующий подход. Прямой праймер (1Re-for– forward) первого экзона содержал фланкирующую область около 15 нуклеотидов и включал сайт рестрикции BamHI, который использован для встраивания в вектрор pET-28a(+). Остальные прямые праймеры (1Re-for–7Re-for) включали две части. Первая часть содержала последовательность (20 ± 3 нуклеотидов) конца предыдущего экзона и служила связующим звеном между экзонами (область перекрывания, см. рис.1). Вторая часть праймера состояла из 20 ± 3 нуклеотидов начала амплифицируемого экзона. Длину праймера подбирали в зависимости от A-T/G-C состава последовательности ДНК. Обратные праймеры (Re-rev – reverse) конструировали (20 ± 3 нуклеотидов) так, чтобы они были комплементарны последовательности концевой части амплифицируемого экзона. Праймер последнего экзона (7Re-rev) содержал фланкирующую область (до 15 нуклеотидов), которая включала сайт рестрикции XhoI, необходимый для встраивания в плазмидный вектор pET-28a(+) (табл. 1).
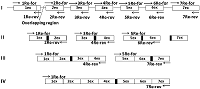
Синтез кДНК гена реналазы человека проводили на матрице ДНК человека, полученной из лимфоцитов цельной крови. Полную кодирующую область гена реналазы собирали в 4 этапа (рис. 1).
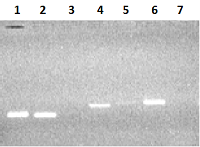
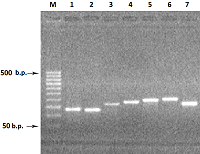
На первом этапе на матрице ДНК человека методом ПЦР получали семь единичных экзонов. При амплификации одиночных экзонов обычно получаются минорные полосы фрагметов ДНК экзонов с хорошим выходом продукта ампликонов в реакции. Однако при амплификации экзонов 3, 5 и 7 синтезировались фрагменты ДНК экзонов с малым выхода ПЦР продукта (рис 2).
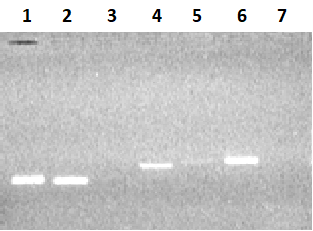
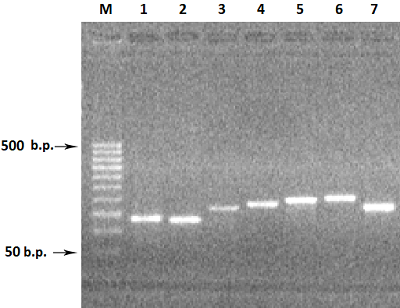
Для увеличения выхода продукта амплификации, при амплификации 3-го экзона увеличивали концентрацию матрицы геномной ДНК в 3 раза (150 нг ДНК), а при реамплификации 5-го и 7-го экзона в качестве матрицы использовали ампликоны от первой амплификации (1-2 мкл реакционной смеси). В результате амплификации с матрицы ДНК семи экзонов в раздельных ПЦР и последующей реамплификации экзонов 3, 5 и 7 были получены ампликоны ДНК экзонов: 1Re - 131 п.о., 2Re – 126 п.о., 3Re – 166 п.о., 4Re – 179 п.о., 5Re – 193 п.о., 6Re – 197 п.о., 7Re – 172 п.о. (рис. 3).
На втором этапе методом ПЦР проводили парное объединение экзонов 1Re со 2Re, 3Re с 4Re и 5Re с 6Re. Матрицей служили ампликоны ДНК одиничных экзонов, а праймерами для первой пары - 1Re-for и 2Re-rev; для второй пары – 3Re-for и 4Re-rev; третьей пары – 4Re-for и 4Re-rev. Связующим звеном при объединении экзонов служила область перекрывания (~20 нуклеотидов), которая была общей для экзонов, т.е. концевая область начального экзона соответствовала начальной области следующего экзона. В результате амплификации трех раздельных ПЦР были получены три фрагмента ДНК парных экзонов: 1Re-2Re – 236 п.о, 3Re-4Re - 325 п.о., 5Re-6Re – 370 п.о. (рис. 1, 4a).
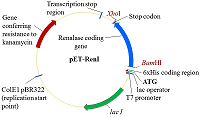
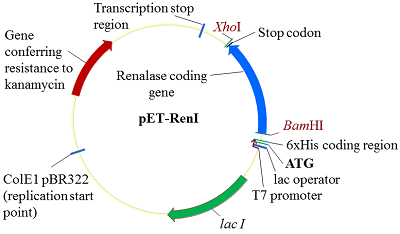
На третьем этапе объединяли парные экзоны: 1Re-2Re с 3Re-4Re и 5Re-6Re 7Re1. В качестве матрицы были использованы ампликоны парных экзонов от второго этапа, а праймерми служили 1Re-for для первой ПЦР и 4Re-rev и 5Re-for и 7Re-rev для второй ПЦР. В результате амплификации двух раздельных ПЦР были получены 2 фрагмента ДНК объединенных 4-х экзонов и 3-х экзонов: 1Re-4Re – 538 п.о, 5Re-7Re – 535 п.о. (рис. 4b). На четвертом заключительном этапе синтеза полноразмерной кДНК гена реналазы человека объединяли фрагменты ДНК четырех и трех экзонов - 1Re-4Re с 5Re-7Re. В качестве матрицы использовали ампликоны ДНК экзонов от третьего этапа, а праймерми служили 1Re-for и 7Re-rev. В результате синтеза был получен фрагмент ДНК 1Re-7Re – 1061 п.о., фланкирующие концы прямого и обратного праймеров содержали сайты рестрикции BamHI и XhoI, соответственно (рис. 4c). Клонирование и экспрессия Конечный ПЦР продукт сборки экзонов - фрагмент ДНК размером 1061 п.о. - вначале очищали в 2% агарозном геле, а затем проводили выделение и очистку с помощью системы для очистки фрагментов ДНК (Wizard), согласно протоколу производителя. Очищенный таким образом фрагмент ДНК был встроен в плазмиду pGEM-T. В результате рестриктного анализа были отобраны 5 клонов. После секвенирования клонированной последовательности ДНК и проведения BLAST анализа (программное обеспечение (National Center for Biotechnology Information, Bethesda, MD, USA https://blast.ncbi.nlm.nih.gov/) был выбран вариант гена реналазы, который соответствовал нуклеотидной последовательности гену реналазы человека банка данных (код доступа GenBank www.ncbi. Homo sapiens NM_001031709). Эта последовательность была вырезана по сайтам рестрикции BamHI/XhoI и переклонирована в вектор pET-28a(+). При встраивании кДНК последовательности в вектор pET-28a(+) ген реналазы находился под контролем Т7-промотора и Lac-оператора (рис. 5).
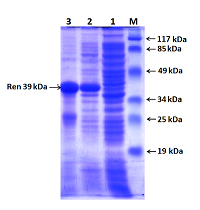
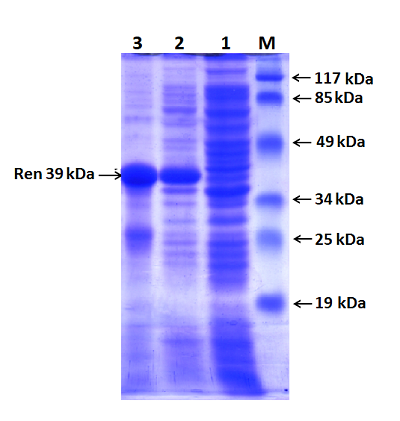
Полученный экспрессионный вектор был обозначен как pET-RenI. Экспрессию проводили в клетах E.coli Rosetta (DE3). В результате экспрессии был получен белок, который на N-конце содержал 6 гистидиновый пептид (6xHis), необходимый для очистки белка на колонке с Ni-Sepharose. Экспрессированный белок анализировали электрофоретически в 10 % ПААГ (рис. 6). По расчетным данным, белок реналазы содержит 342 аминокислотных остатка (а.о.), включал 6 гистидинов и соответствовал молекулярной массе 39 кДа.
ОБСУЖДЕНИЕ При амплификации фрагментов ДНК одиночных экзонов используется матрица геномной ДНК человека, выделенной из лимфоцитов крови. Выделение геномной ДНК является рутинной процедурой и не занимает много времени. Если амплификация экзона дает незначительный выход целевого продукта, то повторная амплификаци ДНК (реамплификация) экзонов дает хороший выход целевого продукта. Так, в нашем случае, при амплификации с матрицы геномной ДНК 3-й и 7-й экзоны при электрофорезе в агарозном геле не выявлялась видимая полоса целевого ампликона или получали низкий выход ПЦР продукта 5-го экзона (рис. 2). В этом случае на первом этапе амплификации экзонов либо увеличивали в 2-3 раза концентрацию матрицы геномной ДНК (при амплификации экзона 3), либо проводили реамплификацию (в случае амплификации экзонов 5 и 7). Реамплификацию проводили в тех же условиях за исключением того, что вместо матрицы геномной ДНК использовали ампликоны ДНК от ПЦР (1-2 мкл), а количество циклов уменьшали до 30. При амплификации ДНК объединяемых экзонов в качестве матрицы используется 0.5-1 мкл ПЦР смеси ДНК ампликонов экзонов без предварительной их очистки, что значительно упрощает и ускоряет процедуру следующего этапа амплификации. Незначительная примесь праймеров или ампликоны от предыдущей амплификации не влияет на амплификацию ДНК при объединении экзонов и не дает побочные ПЦР-ампликоны. Это свидетельствует о том, что процесс амплификации главным образом осуществляется на ДНК ампликонах, добавляемых в качестве матрицы. Другими авторами [10, 11] также описан метод получения полной кодирующей последовательности гена посредством объединения единичных экзонов полученных с матрицы геномной ДНК методом ПЦР. Однако в первой работе используется двухэтапная амплификация при синтезе каждого экзона с обязательной очисткой ампликонов от каждой ПЦР, а во второй работе - трудоемкая процедура очистки ампликонов и их анализ на нативность нуклеотидной последовательности. В нашем случае, при клонировании полной последовательности кДНК гена реналазы, идентичность нуклеотидной последовательности кДНК анализировали только на конечном этапе сборки гена. Полную кДНК последовательность гена реналазы клонировали вначале в вектор pGEM-T. В результате рестриктного анализа (BamHI/XhoI) клонов в рекомбинантной плазмиде pGEM-T выявляли фрагменты ДНК размером 1060 п.о. По данным секвенирования кДНК гена реналазы в рекомбинантных плазмидах, выделенных из пяти отобранных клонов, в нуклеотидной последовательности гена реналазы 1-го клона были обнаружены три точечные мутации в положении 107, 342 и 723. Однако эти мутации затрагивали третий нуклеотид вырожденных триплетов и не меняли кодируемые аминокислоты. Ген реналазы клона 2 и клона 4 имел мутации, затрагивающие аминокислотную последовательность белка. Ген реналазы 3-го и 5-го клона не содержал точечных мутаций. Исходя из этих данных можно сделать вывод, что в клонах, содержащих рекомбинантный ген реналазы, с высокой вероятностью можно отобрать последовательность, которая соответствует нуклеотидной последовательности гена реналазы человека из базы банка данных (код доступа GenBank www.ncbi. Homo sapiens NM_001031709). Следует отметить, что сама ПЦР может вводить мутации в амплифицированную ДНК. Такие мутации являются результатом недостаточно высокой репликативной точности работы Taq ДНК-полимеразы. Для понижения мутационного давления обычно уменьшают общее количество циклов в амплификации ДНК, либо используют термостабильную ДНК полимеразу с корректирующей активностью [9, 12, 20]. Для наших целей мы использовали Tersus-ДНК полимеразу («Евроген»), представляющую собой, специально разработанную смесь термостабильных ДНК полимераз для эффективной амплификации длинных фрагментов ДНК и обладающей корректирующей 3' → 5' экзонуклеазной активностью. Суммируя результаты, полученные при помощи предлагаемого нами «экзонового» метода клонирования рекомбинантного гена реналазы, следует отметить следующие преимущества данного метода: – данный подход является универсальным и простым методом при клонировании рекомбинантных генов, который не требует значительных временных и материальных затрат в отличие от известных в настоящее время методик. – алгоритм подбора праймеров прост и производится исходя из банка данных нуклеотидной последовательности геномной ДНК, а число праймеров равно удвоенному числу экзонов клонируемого гена. – для данного метода не требуются клетки и ткани животных или человека в которых повышена экспрессия клонируемого гена. Использование ткани человеческого материала может быть сопряжено (например, в случае посмертного материала) с соблюдением юридически установленных процедур. Данный подход клонирования генов мы использовали при клонировании гена реналазы-1 и -2 [21, 22], а также при клонировании различных форм реналазы в плазмидные вектора pET-28 и pcDNA4 (Fedchenko et al., in preparation). ФИНАНСИРОВАНИЕ РАБОТЫ Работа выполнена в рамках Программы фундаментальных научных исследований государственных академий наук на 2013–2020 годы и частично поддержана грантом РФФИ № 17–04–00484 (2017–2019). СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Здоровые добровольцы дали информированное согласие на использование их образцов крови в исследовательских целях. ЛИТЕРАТУРА
|