|
Фильтрация данных 2D электрофореза при создании выборки для предсказания значения изоэлектрической точки белков
Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, Ключевые слова: изоэлектрическая точка; 2D электрофорез; подбор данных DOI: 10.18097/BMCRM00162 ВВЕДЕНИЕ
Ранее мы представили программу предсказания изоэлектрической точки (pI) пептидов и белков [1], основанную на использовании уравнения Хендерсона-Хассельбаха [2]. При этом табличные значения pKa были рассчитаны на основании данных, полученных для большой выборки пептидов, в том числе и с различными химическими и посттрансляционными модификациями (PTM), в экспериментах по изоэлектрофокусированию пептидов с последующей масс-спектрометрической идентификацией. В свою очередь, эта выборка пептидов была получена в результате тщательной фильтрации полного набора данных многократно большего размера, проведённой с целью увеличения процента достоверных значений в выборке. В то же время тестирование качества предсказания pI для белков показало, что качественного большого набора данных для решения этой задачи нет. Отдельные выборки, полученные в экспериментах по двумерному (2D) гель-электрофорезу, имеют, на первый взгляд, хорошее качество и предсказываются хорошо. Однако в случае смешанных выборок, например из работы [3], около 60% наблюдений имеют ошибку предсказания не хуже 0.5 значений pH, в пределах ошибки до 0.1 значений pH было всего 15% [1]. Для решения некоторых задач такой точности могло бы хватить, но для идентификации конкретной протеоформы [4] этого оказывается недостаточно. Возникает вопрос, является ли невысокая точность предсказания проблемой самого метода предсказания? На выборках, полученных в рамках одного эксперимента, результаты были значительно лучше, как в работе [1], так и при использовании других методов, основанных на применении уравнения Хендерсона-Хассельбаха, в том числе и использованного далее в настоящей работе [5]. Почему возникают сомнения в качестве подобных сборных выборок? В первую очередь, этот результат отражает стремительный рост всевозможных омик-наук и направления «big data». Данные становятся вторичными и теряют часть информации, содержавшейся в первоисточниках. Попадая в различные базы данных, эти вторичные данные становятся основой для формирования новых выборок и это может быть не одна итерация. В области предсказания значения pI для белков примером такой вторичной базы данных может быть Proteome-pI [6]. При этом, на каждом этапе исследователи решают свои собственные задачи, и часто для них часть информации несущественна, но в случае, когда идёт речь о «big data», достоверность данных имеет ключевое значение [7]. В настоящей работе показаны некоторые «подводные камни», которые могут свести на нет достоверность выборок при автоматической обработке исходных данных. МЕТОДИКА Для анализа данных были использованы 2 различных варианта предсказания значения pI для белков, базирующиеся на использовании уравнения Хендерсона-Хассельбаха. Первый из них, разработанный Bjellqvist и соавт. [5] – самый часто применяемый в настоящее время, так как именно он используется в программе предсказания pI с сервера expasy.org. Второй, разработанный нами [1], кроме собственной шкалы pKa, охватывающей не только основные аминокислотные остатки, но и остатки с модификациями, различает также местоположение аминокислотного остатка в полипептидной цепи. Для немодифицированных аминокислотных остатков в целом на больших выборках оба метода дают близкие результаты [1], но в каждом конкретном случае могут быть некоторые различия. В связи с этим для надёжности и внутреннего контроля лучше использовать оба. В качестве демонстрационных выборок данных для белков с «известными» значениями pI были использованы несколько наборов данных, полученных в экспериментах по 2D гель-электрофорезу (табл. 1), депонированных в World-2DPAGE Repository [8]. Данные были собраны собственной программой, которая также автоматически подгружала последовательности идентифицированных белков из БД Uniprot [9]. Если автоматизированная процедура не смогла найти аминокислотную последовательность в БД, данное наблюдение отбрасывали. Как правило, это связано с тем, что в различных редакциях БД идентификаторы были изменены, объединены или удалены (нельзя исключить также и факт ошибок и опечаток при формировании страницы в World-2DPAGE Repository.) Используя архивные данные, можно отыскать вручную все измененные последовательности, но так как задачей нашей работы было показать именно проблемы автоматизированных процедур, этого не делали. Для анализа данных использовали координаты (номера пикселей по горизонтали), поставленные в соответствие точке идентификации, приписанное значение pI и предсказанные значения pI.
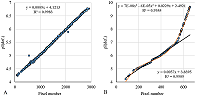
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ При анализе результатов из любых глобальных БД всегда следует помнить, что они вторичны и сбор информации или её ввод авторами осуществляется либо вручную, либо с использованием внешних программ. И в том, и другом случае возможны ошибки ввода. Автоматизированные процедуры не предполагают работы оператора со статьями, где были опубликованы первичные данные, а в случае работы с большими данными («big data») это физически невозможно. Если информация, содержащаяся в изначальной статье, не соответствует структуре изначальной публикации, то она теряется или, что ещё хуже, искажается. Тем не менее, возможность проанализировать данные на ошибки или выявить отдельные особенности, которые следует учесть при отборе информации, все-таки существует. Рассмотрим два набора данных RAT_INS1E_4-7 и RBC_HUMAN (рис. 1). Если проанализировать значение pI и координаты по оси абсцисс на опубликованной карте (проще говоря, номер пикселя по горизонтали), то из общих соображений ясно, что зависимость должна быть монотонной. В зависимости же от типа «стрипа» (полоска с иммобилизованным градиентом рН), использованного в эксперименте, эта зависимость может быть либо линейной, либо соответствующей заявленной производителем (а их не так много). Данные RAT_INS1E_4-7 (рис. 1A) явным образом указывают на линейную зависимость, однако имеется несколько точек (2 из них ярко выражены), отклонение которых нельзя описать ошибками округления. С 100% вероятностью эти точки можно считать ошибками. Второй случай (рис. 2B), на первый взгляд, соответствует нелинейной зависимости pI от координат, которая может быть описана кубическим сплайном и не имеет видимых ошибок. Характер кривой напоминает ожидаемую зависимость, такую как, например, у Nonlinear pH 3-10 ReadyStrip IPG strip («Bio-Rad Laboratories, Inc.», США). В тоже время при автоматизированной обработке данных сложно учесть все варианты, если этих данных нет в БД. Тем более, что предсказанные величины pI (например, методом Bjellqvist, см. дополнительные материалы) часто не совпадают с наблюдаемыми, сохраняя линейность и в этой части. Хотя шкала, полученная Bjellqvist и соавт. ещё в 1983 году, основана на данных для диапазона pI от 4 до 7, другие методы также дают близкие результаты. При первичной фильтрации данных пользоваться результатами каких-либо предсказаний неправильно. Существующий выход – вычленять данные для линейного участка (рис. 1 B) и ограничиться ими. Для 2D электрофоретических карт редко, но встречается и ещё один вариант, когда авторы просто приписывают конкретным белкам «теоретические» значения pI. Поскольку при этом для расчёта используется программа с сервера expasy.org, то простое сравнение данных с результатами предсказания Bjellqvist и соавт. легко выявляет такой случай, обычно это видно и при анализе зависимости pI от координат на карте.
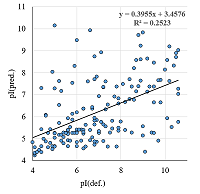
Отдельно необходимо рассмотреть случай, когда исследуются белки организмов, для которых неизвестен, либо частично известен геном, например, белки морской звезды Marthasterias glacialis [16]. В такой ситуации при масс-спектрометрической идентификации используют последовательности близких ли нет видов. Ожидать, что в этом случае совпадут значения pI, полученные из 2D электрофоретической карты, и предсказанные величины не приходится (рис. 2). В рассматриваемом примере в списке нет ни одного белка Marthasterias glacialis. Если добавить в процедуру сбора данных простое сравнение по видовой принадлежности, то это решит проблему. К сожалению, в случае внутривидовых различий, особенно выраженных у одноклеточных организмов, это не поможет. В данном случае можно только надеяться, что один и тот же функционально белок имеет и схожие физико-химические свойства.
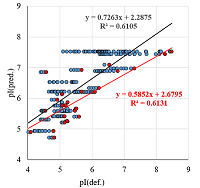
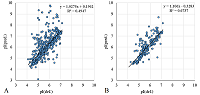
Ещё одним фактором, вносящим существенные искажения в значения pI, является наличие PTM. В 2D электрофорезе это выглядит как идентификация одного белка для нескольких «пятен». Широко распространённой практикой среди исследователей, формирующих выборки для создания методов предсказания значений pI, является усреднение значений [3]. В ряде случаев, если варианты мало различаются по величине pI, это не вносит больших искажений. Но пример, представленный на рисунке 3, демонстрирует обратное. Статистически усреднённые данные схожи с данными, полученными при отборе конкретного значения, но полученные зависимости существенно отличаются. Возникает вопрос, каким образом выбрать конкретное значение? Универсального решения нет. Из общих соображений это может быть самое большое по площади пятно на 2D карте, но данной информации в БД может и не быть. Более приемлемый вариант – выбирать самое оснóвное значение pI, так как большинство имеющихся физиологических PTM смещают значение pI в кислую сторону. Одна PTM всё же может быть учтена. Речь идёт об удалении N-концевого остатка метионина. В зависимости от объекта исследований решение о сохранении его при расчётах или удалении может быть принято заранее. И если для такого метода предсказания, как Bjellqvist и соавт., эта процедура не даст никаких изменений, то предложенный нами метод предсказания pIPedict [1] и ряд других учтут это изменение. Все предсказанные величины до этого момента были получены методом Bjellqvist и соавт. – не самым лучшим, но самым распространённым и известным среди исследователей в области 2D электрофореза. Однако предложенный нами метод предсказания pIPredict дает несколько лучший результат (рис. 4), поэтому в дальнейшем все предсказания были выполнены этим методом.
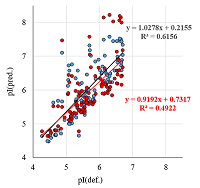
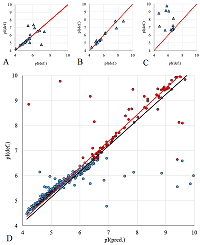
Хорошим вариантом при формировании выборки значений pI было бы подтверждение значения как минимум в двух экспериментах. Однако выборка при этом может быть очень небольшой. В нашем случае (рис. 5 A,B) для всех 5 карт Homo sapiens как минимум 2 значения было всего у 31 белка, в двух картах Mus musculus имеется 17 совпадений. И даже в этом случае не все приписанные значения pI совпадают (при наличии вариантов использовали самое основное значение). На рисунке 5 С приведены данные для пересечения двух карт Staphylococcus aureus, выполненных одними и теми же авторами в двух различных диапазонах pH, имеющих пересечение в области значений от 6 до 7. Для 8 из 19 пересекающихся белков приписанные значения отличаются кардинально. Кроме того, если сравнить приписанные значения с предсказанными (рис. 5 D), то хорошо видно, что для правой части имеет место небольшое смещение вверх. Если ориентироваться на приписанные величины pI для пересекающихся белков, то это смещение примерно в 0.1-0.15 значений pH. Величина небольшая, но вполне значимая.
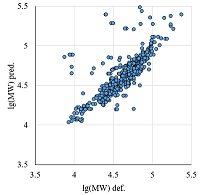
Ну и наконец, до сих пор никак не обсуждался второй параметр из 2D карт – электрофоретический сдвиг, пропорциональный молекулярному весу белков (MW). Этот параметр также зависит от множества факторов: тех же PTM, гликозилирования (на величину pI практически не влияет), наличия в пробах редуцированных форм или фрагментов белков, наличия в составе белков аминокислотных паттернов, характерных для устойчивых элементов вторичной структуры и не денатурирующих полностью [17], различиями в способности конкретных белков связывать молекулы SDS (т.е. в конечном итоге аминокислотный состав и те же паттерны) и др. К сожалению, в большинстве случаев в настоящее время нет возможности без знания всех особенностей исследуемого белка определить, почему в электрофоретическом сдвиге наблюдаются отклонения от ожидаемого. Для каких-то конкретных модельных белков с заранее точно известными аминокислотной последовательностью и имеющимися модификациями можно найти данные по электрофоретическому сдвигу, но в подавляющей части случаев при анализе природных проб точных данных нет. Тем не менее, можно предположить, что если величина отклонения от ожидаемого велика, то мы имеем дело с какой-то формой, отличной от простой полной аминокислотной последовательности из БД Uniprot. Как видно из рисунка 6, в основном различия между расчётной величиной MW и наблюдаемой в большинстве случаев находятся в пределах 0.2 единиц lg(MW). Большее отклонение можно заведомо считать ошибкой. Предел отклонения по MW может быть и более жёстким. При этом число наблюдений в выборке будет уменьшаться, но не катастрофично, а качество выборки будет расти (рис. 7). Если при первоначальной фильтрации (на явные ошибки, выбору линейного участка зависимости
ЗАКЛЮЧЕНИЕ Таким образом, при применении очень простых фильтров, установленных из общих соображений, учитывающих особенности проведения экспериментов и природу полученных результатов, качество автоматически собранных выборок для обучения систем предсказания значений pI белков может быть существенно улучшено. Число таких фильтров не исчерпывается теми, что описаны в данной статье. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Данная работа не содержит каких-либо исследований с использованием людей и животных в качестве объектов исследования. ФИНАНСИРОВАНИЕ Работа выполнена в рамах Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021 - 2030 годы) (№ 122030100170-5).
К данной статье приложены дополнительные материалы, свободно доступные
(http://dx.doi.org/10.18097/BMCRM00162) на сайте журнала.
ЛИТЕРАТУРА
|