|
Простая методика выделения плазмид из Escherichia Coli для эффективной химической трансфекции культуры клеток человека Федеральный научно-клинический центр физико-химической медицины федерального медико-биологического агентства, 119435, Москва, ул. Малая Пироговская, 1а; *e-mail: lazarev@rcpcm.org Ключевые слова: бактериальные плазмиды; трансфекция; линия клеток человека DOI: 10.18097/BMCRM00170 ВВЕДЕНИЕ Трансфекция клеточных линий человека плазмидами, полученными из Escherichia coli, является широко распространенным методом, применяемым для решения различных исследовательских и биотехнологических задач [1, 2]. При этом, для эффективной трансфекции качество плазмидной ДНК (пДНК) должно быть достаточно высоким. Во-первых, пДНК должна быть хорошо очищена от примесей, в первую очередь от бактериальных липополисахаридов [3] и белков [2]. Во-вторых, основная часть плазмид должна находиться в форме с отрицательной сверхспирализацией [4]. Таким образом, при выделении пДНК, с одной стороны, необходимо проводить ряд последовательных стадий для удаления примесей, часто в достаточно жестких условиях, а с другой – избегать повреждений и разрывов сахарофосфатного остова ДНК, что ведет к релаксации суперскрученной формы. В подавляющем большинстве случаев первой стадией выделения пДНК является щелочной лизис [5, 6]. пДНК, полученная после щелочного лизиса и осаждения спиртом, пригодна для ряда задач, таких как ПЦР, рестрикция, секвенирование и т. п. Однако для трансфекции требуется проводить дополнительные стадии очистки. Самыми распространенными методами очистки являются фенол-хлороформовая экстракция, связывание с частицами диоксида кремния, анионобменная хроматография, гель-фильтрация. Для получения пДНК с чистотой, соответствующей фармокологическим стандартам, применяют сложные схемы хроматографической очистки со специальными сорбентами [7]. Мы предлагаем простую лабораторную методику выделения плазмидной ДНК, состоящую из трех стадий: щелочного лизиса, связывания с диоксидом кремния и гель-фильтрации. Методика подходит для получения пДНК для трансфекции. Мы показали, что эффективность трансфекции с использованием пДНК, выделенной по описываемой методике, не уступает эффективности трансфекции с использованием пДНК, выделенной коммерческим набором Qiagen plasmid maxi kit ( «Qiagen», США). МАТЕРИАЛЫ И МЕТОДЫ Реактивы В работе использованы: ЭДТА, NaCl, NaOH, додецилсульфат натрия, Tris-base, ацетат натрия («Реахим», Россия); дрожжевой экстракт, триптон, ампициллин, агароза, бромистый этидий («Хеликон», Россия); частицы оксида кремния S5631 («Sigma», США), суспензия 300 г/л в 10 мМ TrisCl, 1 мМ ЭДТА, pH 7,5; фетальная сыворотка крупного рогатого скота, гентамицин, DMEM, OptiMEM, HBSS («Life Technologies», США); Lipofectamine™ 3000 Transfection Kit («Invitrogen», США); хроматографический сорбент Sephacryl S-500 HR («Cytiva», США). Растворы Раствор I: 20 mM TrisHCl pH 7.5, 1 мM ЭДТА, сахароза 200 г/л; раствор II: 10 г/л SDS, 0.2 М NaOH; раствор III: 3 М CH3COONa , 2М CH3COOН; раствор W: 100 mM TrisHCl pH 7.5, 50 мМ NaCl, 1 мM ЭДТА, 50% (об.) этанол; раствор Е: 20 mM TrisHCl pH 7.5, 1 мM ЭДТА, 10 мг/л РНКаза А («Sigma», США); PBS: 20 мМ фосфат натрия, 8 г/л NaCl, pH 7.4; бактериальная среда LB: триптон 10 г/л, дрожжевой экстракт 5 г/л, NaCl 10 г/л. Клетки Штамм E. coli Top10: F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15; линия клеток человека Expi293F («Thermo Fisher Scientific», США). Оборудование Хроматограф AKTA Start («GE Healthcare», США); спектрофотометр UV-1900 («Shimadzu», Япония); CO2-инкубатор Heraeus Heracell 240 («Thermo Fisher Scientific», США); проточный цитофлуориметр NovoCyte Flow Cytometer («ACEA Biosciences», США). Культивирование бактерий В работе использовали штамм E. coli Top10, трансформированный плазмидой pEGFP-N1 («Clontech», США). Единичную колонию клеток переносили в культуральную колбу объемом один литр, содержащую 100 мл среды LB с канамицином (50 мг/л). Колбу помещали в шейкер-инкубатор и инкубировали 16 ч при температуре 37°С и 240 об/мин. Клеточную культуру переносили в центрифужный стакан и центрифугировали в течение 10 мин при 5000 g и 4°С. Осадок ресуспендировали в 50 мл PBS и повторно центрифугировали в тех же условиях. Надосадочную жидкость сливали, а клеточный осадок замораживали и хранили при -20°С. Щелочной лизис Центрифужную пробирку, содержащую замороженный осадок бактерий, оттаивали на льду. К осадку добавляли 10 мл раствора I и тщательно ресуспендировали. Далее, к суспензии добавляли 10 мл раствора II и перемешивали десять раз, переворачивая пробирку, после чего помещали на лед на 5 мин. К лизату добавляли 15 мл раствора III перемешивали десять раз, переворачивая пробирку, после чего помещали на лед на 5 мин. Выпавший осадок отделяли с помощью центрифугирования при 4°С и 15000 g в течение 15 мин. Полученный раствор объемом 35 мл переносили в новую центрифужную пробирку. Связывание пДНК с частицами оксида кремния К раствору, полученному на предыдущей стадии, добавляли 15 мл 6 М раствора гуанидинхлорида и 0.5 мл суспензии частиц диоксида кремния. Пробирку перемешивали переворачиванием и инкубировали при комнатной температуре 15 мин, периодически перемешивая. Частицы диоксида кремния осаждали центрифугированием 1 мин при 5000 g. Супернатант удаляли, а осадок ресуспендировали в 10 мл 6 М гуанидинхлорида и центрифугировали 1 мин при 5000 g. Супернатант удаляли, а осадок ресуспендировали в 25 мл раствора W и центрифугировали 1 мин при 5000 g, после чего повторяли отмывку раствором W. Осадок высушивали 10 мин под вакуумом и размешивали в 2 мл раствора E. Суспензию инкубировали 15 мин при 37°С, периодически перемешивая. Частицы диоксида кремния осаждали центрифугированием 5 мин при 5000 g, раствор пДНК переносили в новую пробирку. Гель-фильтрация Разделение проводили на хроматографической колонке Tricorn 10/100 10х50 мм («Cytiva»), заполненной сорбеном Sephacryl S-500 HR. Колонку уравновешивали PBS. К 1 мл раствора пДНК, полученному н предыдущем этапе, добавляли 10 мкл раствора SDS (100 г/л) и наносили на колонку со скоростью потока 1 мл мин. Контроль разделения осуществляли на основе проточного измерения оптической плотности при 280 нм. пДНК выходила в свободном объеме колонки, в то время как более низкомолекулярные примеси сходили с задержкой. К фракции, содержащей пДНК, добавляли равный объем изопропилового спирта, 1/10 объема раствора III, инкубировали 10 мин при -20°С и центрифугировали при 15000 g в течение15 мин. Супернатант удаляли, осадок высушивали под вакуумом и растворяли в 0.5 мл воды. Перед повторным использованием колонку промывали 10 мл 0.5 М NaOH и уравновешивали PBS. Определение эффективности трансфекции Эффективность трансфекции определяли по флуоресценции белка eGFP в эукариотических клетках эмбриональной почки человека Expi293F. Для этого использовали транзиентную трансфекцию этих клеток плазмидными конструкциями, описанными выше, с использованием трансфецирующего агента Lipofectamine™ 3000. Накануне трансфекции клетки Expi293F рассевали в 48-луночный планшет из расчёта 5х104 клеток в лунку в 1 мл среды DMEM, содержащей 10% фетальной сыворотки крупного рогатого скота и гентамицин 10 мкг/мл. Клетки инкубировали в СО2-инкубаторе в атмосфере 8% СО2. На следующий день готовили смеси реагента LipofectamineTM 3000 со средой OptiMEM из расчета 0.75 мкл реагента на 50 мкл среды. LipofectamineTM 3000 реагент добавляли непосредственно в среду. Сразу после внесения реагента перемешивали аккуратно полученную суспензию двукратным пипетированием. Затем разводили 1 мкг плазмидной ДНК в 50 мкл среды OptiMEM, перемешивали пипетированием. К раствору ДНК добавляли 1 мкл реагента P3000TM, инкубировали раствор 5 мин. К готовому раствору ДНК с реагентом P3000TM по каплям добавляли разведённый реагент LipofectamineTM 3000. Раствор аккуратно пипетировали два раза для равномерного перемешивания. Инкубировали смесь 10-15 мин при комнатной температуре. После этого по каплям добавляли суспензию комплексов ДНК/ LipofectamineTM 3000 к клеткам в каждую лунку ранее приготовленного культурального планшета. Аккуратно перемешивали планшет круговыми движениями. Через 48 ч после начала трансфекции отбирали аккуратно культуральную среду, суспендировали клетки в 200мкл буфера HBSS. Эффективность трансфекции оценивали на проточном цитофлуориметре NovoCyte Flow Cytometer с использованием программного обеспечения The NovoExpress software («ACEA Biosciences»). Определение концентрации пДНК. Концентрацию ДНК определяли по оптической плотности при длине волны 260 нм. Измерения проводили с помощью спектрофотометр UV-1900 («Shimadzu», Япония) в кварцевых кюветах с длиной оптического пути 1 см. При пересчете использовали коэффициент экстинкции A0,1%=20. Раствор пДНК разводили водой в 20 раз, при этом значение ОП260 численно равнялось концентрации исходного раствора ДНК в мг/мл. Дополнительно измеряли оптическую плотность при длине волны 280 нм и рассчитывали отношение ОП260/ОП280, как один из критериев чистоты образца ДНК. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Выделение пДНК Для получения образцов сравнения помимо пДНК, выделенной по полной описаной методике (образец 1), были получены образцы пДНК, выделенные по методике с исключением отдельных стадий. В первом случае после щелочного лизиса и очистки с помощью диоксида кремния, пДНК осаждали изопропанолом и растворяли в воде, а стадию гель-фильтрации не проводили (образец 2). Во втором случае исключали стадию связывания с частицами диоксида кремния (образец 3). Раствор после щелочного лизиса осаждали изопропиловым спиртом, растворяли в растворе Е и переходили сразу к стадии гель-фильтрации. Для того, чтобы исключить флуктации при наращивании биомассы бактерий, перед цетрифугированием содержимое четырех культуральных колб объединяли, перемешивали, потом снова делили на четыре части, а затем осаждали клетки центрифугированием. Далее из каждой части выделяли пДНК одним описаным выше способов. Кроме того, из одной части проводили выделение пДНК с помощью комерческого набора Qiagen plasmid maxi kit (образец 4).
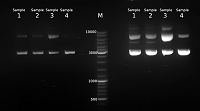
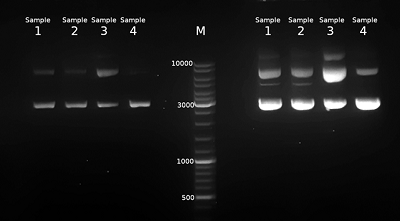
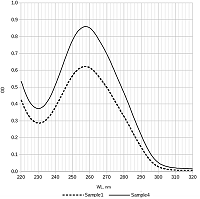
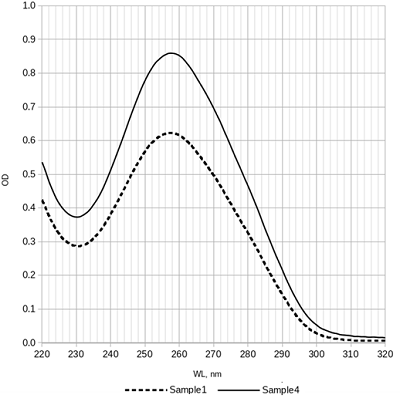
В результате во всех случаях была получена плазмидная ДНК, находящаяся преимущественно в сверхспирализованной форме (рис. 1). При этом, наименьшее количество релаксированной формы наблюдается в образце, выделенном набором Qiagen plasmid maxi kit, а наибольшее — в образце 3, полученном без стадии очистки с помощью диоксида кремния. В количественном отношении наилучший результат был получен в случае образца 3 — 1005 мкг, а наихудший в случае образца 1 — 300 мкг (табл. 1). Можно заключить, что наибольшие потери возникают на стадии очистки с помощью диоксида кремния. Соотношение количества пДНК в образцах 3 и 1 составляет 3.3 раза. В то же время потери при гель-фильтрации не столь велики, соотношение количества пДНК в образцах 2 и 1 составляет 1.2 раза. Соотношение оптической плотности образцов при длинах волн 260 нм и 280 нм для всех четырех образцов хорошее и составляет более, чем 1.8 раза (табл. 1). Спектр поглощения в ультрафиолетовой области имеет характерный для ДНК вид с максимумом при 260 нм (рис. 2).
Эффективность трансфекции Эффективность трансфекции оценивали с использованием проточного цитофлуориметра, подсчитывая количество клеток, в которых наблюдалась флуоресценция зеленого флуоресцентного белка (eGFP). Результаты представлены в таблице 2. Во всех опытных образцах количество трансфицированных клеток составляло около 95%, различия между образцами статистически незначимы. Таким образом, плазмидная ДНК, выделенная по описанной методике, трансфецирует клетки эмбриональной почки человека линии Expi293F столь же эффективно, как и пДНК, выделенная с помощью специализированного набора реагентов фирмы «Qiagen». Предложенная методика примерно вдвое более затратна по времени, но требует для выполнения только самых простых и дешевых реактивов.
ЗАКЛЮЧЕНИЕ Предложена лабораторная методика выделения плазмид из клеток E. coli для последующей трансфекции линии клеток человека. Методика не требует дорогостоящих реактивов и легко масштабируется. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов. ФИНАНСИРОВАНИЕ Работа выполнена в рамках государственного задания ФМБА России по теме «Разработка комплексной схемы терапии лекарственно-устойчивых возбудителей инфекционных заболеваний с применением бактериофагов или их производных в сочетании с антибактериальными препаратами». КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|