Способ поиска лигандов для новых сайтов связывания
1Институт биомедицинской химии им. В.Н.Ореховича Москва, 119121, Погодинская ул., 10; e-mail: veselov@ibmh.msk.su
2Российский национальный исследовательский медицинский университет имени Н.И.Пирогова г. Москва 117997, ул. Островитянова, 1
Ключевые слова: молекулярный докинг, место связывания, модель фармакофора, молекулярная динамика, HSP90, конструирование de novo
DOI:10.18097/BMCRM00200
Анализ белковых структур показывает, что большинство из них имеют потенциальные сайты связывания, которые можно рассматривать как перспективные для поиска новых лигандов. Однако, отсутствие данных о известных лигандах, взаимодействующих с такими сайтами связывания, сильно ограничивают поиску и отбору новых соединений. В данной работе представлен подход для повышения эффективности виртуального скрининга. Данный подход объединяет методы дизайна лигандов de novo, построения моделей фармакофоров, молекулярный докинг, молекулярную динамику, расчет энергий связывания с помощью MM-GBSA. При использовании данного подхода на первом этапе создается виртуальная библиотека потенциальных соединений методом конструирования de novo, с последующим отбором эффективных субструктур и их расположения в сайте связывания. На основе которых разрабатываются модели фармакофоров, которые используются для виртуального скрининга молекулярных баз данных. Отобранные соединения докируются в сайт связывания для проверки их способности размещаться в нем и для оценки совпадения идентифицированных благоприятных фрагментов в том же районе сайта связывания, предсказанном при генерации молекул de novo. Дальнейшую оценку выбранных лигандов можно проводить стандартными методами компьютерного конструирования лекарств. Предлагаемый подход может способствовать эффективному поиску лигандов для новых сайтов связывания.

|
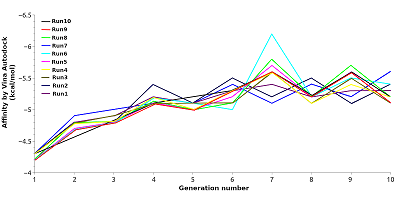
Рисунок 1.
Средние величины предсказанных аффинностей для сгенерированных соединений.
|
|
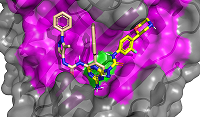
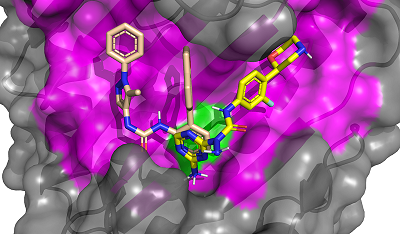
Рисунок 2.
Соединения 1 и 2 в борозде на поверхности HSP90 около остатка Thr-90.
|
|
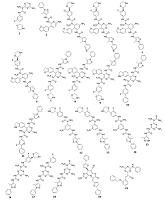
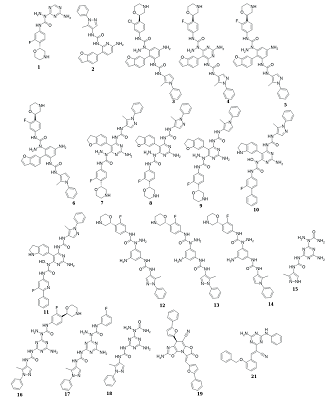
Рисунок 3.
Соединения, использованные для построения фармакофорных моделей.
|
|
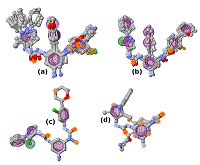
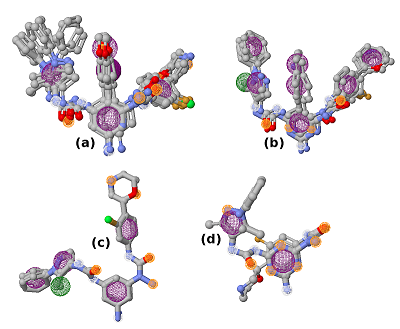
Рисунок 4.
Построенные фармакофорные модели. а – фармакофор 1 (использованные соединения 1-6), b – фармакофор 2 (использованные соединения 7-11), c – фармакофор 3 (использованные соединения 12-14), d – фармакофор 4 (использованные соединения 15-18). Пурпурными сферами показаны ароматические фрагменты, серыми сферами – доноры Н-связей, желтыми сферами – акцепторы Н-связей, зелеными сферами – гидрофобные фрагменты.
|
|
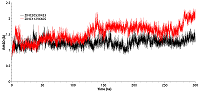
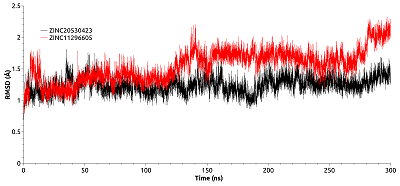
Рисунок 5.
Изменения величин RSMD для соединений 19 (ZINC20530423) и 21 (ZINC1126605) при молекулярной динамике.
|
|
ЗАКРЫТЬ

|
Таблица 1.
Первоначальные фармакофорные модели.
|
|
ЗАКРЫТЬ
|
Таблица 2.
Вторичные фармакофорные модели, использованные для фармакофорного поиска.
|
|
ЗАКРЫТЬ
|
Таблица 3.
Рассчитанные величины энергии связывания комплекса HSP90 с соединениями 19 и 21 методом MM–PBSA.
|
|
ЗАКРЫТЬ
|
Таблица 4.
Рассчитанные величины энергии связывания комплекса HSP90 с соединениями 19 и 21 методом MM–PBSA.
|
ФИНАНСИРОВАНИЕ
Работа поддержана Российским фондом фундаментальных исследований (проект № 19-34-90057).
К данной статье приложены дополнительные материалы, свободно доступные в электронной версии (http://dx.doi.org/10.18097/BMCRM00200) на сайте журнала.
ЛИТЕРАТУРА
- Shin W.-H., Christoffer C.W., Kihara D. (2017) In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods, 131, 22–32. DOI
- Kandel J., Tayara H., Chong K.T. (2021) PUResNet: prediction of protein-ligand binding sites using deep residual neural network. J. Cheminform., 13(1), 65. DOI
- Wang K., Zhou R., Li Y., Li M. (2021) DeepDTAF: a deep learning method to predict protein–ligand binding affinity. Brief. Bioinform., 22(5), bbab072. DOI
- Zhao J., Cao Y., Zhang L. (2020) Exploring the computational methods for protein-ligand binding site prediction. Comput. Struct. Biotechnol. J., 18, 417–426. DOI
- Roche D., Brackenridge D., McGuffin L. (2015) Proteins and their interacting partners: an introduction to protein–ligand binding site prediction methods. Int. J. Mol. Sci., 16(12), 29829–29842. DOI
- Veselovsky A., Archakov, A. (2007) Inhibitors of Protein-Protein Interactions as Potential Drugs. Curr. Comput.-Aided Drug Des., 3(1), 51–58. DOI
- Ni D., Liu Y., Kong R., Yu Z., Lu S., Zhang J. (2022) Computational elucidation of allosteric communication in proteins for allosteric drug design. Drug Discov Today, 27(8), 2226-2234. DOI
- Broomhead N.K., Soliman M.E. (2017) Can We Rely on Computational Predictions To Correctly Identify Ligand Binding Sites on Novel Protein Drug Targets? Assessment of Binding Site Prediction Methods and a Protocol for Validation of Predicted Binding Sites. Cell Biochem. Biophys., 75(1), 15–23. DOI
- Limongelli V. (2020) Ligand binding free energy and kinetics calculation in 2020. WIREs Comput. Mol. Sci., 10, e1455. DOI
- Sink R., Gobec S., Pecar S., Zeg, A. (2010) False Positives in the Early Stages of Drug Discovery. Curr. Med. Chem., 17(34), 4231–4255. DOI
- Awuni Y., Mu Y. (2015) Reduction of False Positives in Structure-Based Virtual Screening When Receptor Plasticity Is Considered. Molecules, 20(3), 5152–5164. DOI
- Culig Z. (2014) Targeting the androgen receptor in prostate cancer. Expert Opin. Pharmacother., 15(10), 1427-1437. DOI
- Thakur A., Roy A., Ghosh A., Chhabra M., Banerjee S. (2018) Abiraterone acetate in the treatment of prostate cancer. Biomed. Pharmacother., 101, 211–218. DOI
- Heinlein C.A., Chang C. (2004) Androgen Receptor in Prostate Cancer. Endocr. Rev., 25(2), 276–308. DOI
- Dagar M., Singh J.P., Dagar G., Tyagi R.K., Bagchi G. (2019) Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J. Biol. Chem., 294(22), 8699–8710. DOI
- Li J., Sun, L.; Xu C., Yu F., Zhou H., Zhao Y., Zhang J., Cai J., Mao C., Tang L., Xu Y., He J. (2012) Structure insights into mechanisms of ATP hydrolysis and the activation of human heat-shock protein 90. Acta Biochim Biophys Sin (Shanghai), 44(4), 300-306. DOI
- Spiegel J.O., Durrant J.D. (2020) AutoGrow4: an open-source genetic algorithm for de novo drug design and lead optimization. J. Cheminform., 12(1), 25. DOI
- Schneidman-Duhovny D., Dror O., Inbar Y., Nussinov R., Wolfson H.J. (2008) PharmaGist: a webserver for ligand-based pharmacophore detection. Nucleic Acids Res., 36, W223–W228. DOI
- Koes D.R., Camacho C.J. (2012) ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res., 40, W409–W414. DOI
- Trott O., Olson A.J. (2009) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem., 31(2), 455-461. DOI
- Adasme M.F., Linnemann K.L., Bolz S.N., Kaiser F., Salentin S., Haupt V.J., Schroeder M. (2021) PLIP 2021: expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res., 49(W1), W530-W534. DOI
- Berendsen H.J.C., van der Spoel D., van Drunen R. (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun., 91(1-3), 43-56. DOI
- Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins, 78(8), 1950-1958. DOI
- Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys., 81(8), 3684–3690. DOI
- Parrinello M., Rahman A. (1980) Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett., 45(14), 1196–1199. DOI