|
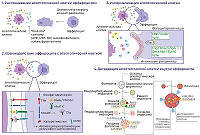
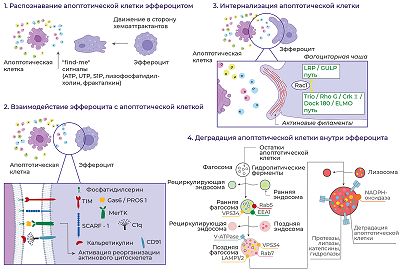
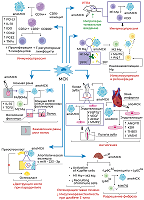
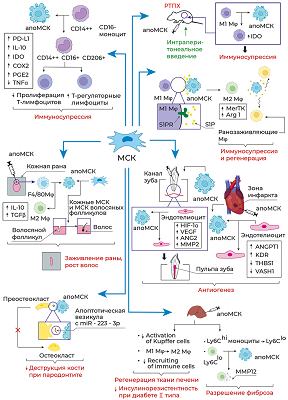
СОДЕРЖАНИЕ 2. БАЗОВЫЕ ХАРАКТЕРИСТИКИ МСК И ИХ ТЕРАПЕВТИЧЕСКИЙ ПОТЕНЦИАЛ 4. ЭФФЕРОЦИТОЗ МСК ОКАЗЫВАЕТ ПРОТИВО-ВОСПАЛИТЕЛЬНЫЕ ЭФФЕКТЫ 5. АПОПТОТИЧЕСКИЕ МСК СПОСОБСТВУЮТ РЕГЕНЕРАЦИИ ТКАНИ И ЗАЖИВЛЕНИЮ КОЖНЫХ РАН СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Рисунок 1Этапы эффероцитоза апоптотической клетки. Описание в тексте. Рисунок 2Механизмы терапевтического действия апоптотических МСК. |
Эффероцитоз как один из механизмов реализации терапевтических эффектов мезенхимных стволовых клеток
Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, Ключевые слова: эффероцитоз; апоптоз; мезенхимные стволовые клетки; воспаление; регенерация; иммуномодуляция DOI: 10.18097/BMCRM00221 ВВЕДЕНИЕ Мезенхимные стволовые (или стромальные) клетки (МСК) обладают набором свойств, делающих их очень привлекательным материалом при разработке технологий клеточной терапии широкого круга болезней. Однако использование МСК в клинике сдерживается недостаточно чётким пониманием клеточных и молекулярных механизмов их терапевтического действия. При этом количество опубликованных данных огромно и их углублённый анализ может существенно улучшить ситуацию. В данном обзоре мы рассматриваем роль апоптоза МСК в реализации их терапевтических эффектов с акцентом на финальную стадию этого варианта клеточной гибели – эффероцитоз. Апоптоз – одна из форм программируемой клеточной гибели – имеет огромное физиологическое значение. На протяжении всей жизни, начиная с эмбриональной стадии и до старения, апоптоз является неотъемлемым компонентом развития и поддержания гомеостаза организма. Апоптоз исключительно важен для морфогенеза, удаления клеток, повреждённых вследствие химического, физического или механического воздействия, генетически дефектных и потенциально опасных трансформированных и/или аутореактивных клеток, а также для поддержания нормального количества клеток в тканях [1]. Например, у эмбрионов позвоночных апоптоз участвует в развитии нервной системы и сенсорных органов [2-4]. Классическим примером участия в морфогенезе является апоптоз клеток межпальцевых перепонок с целью формирования отдельных пальцев [1]. Физиологический апоптоз, происходящий в незрелых семенниках, необходим для созревания ткани, а его ингибирование сопровождается накоплением сперматогоний и бесплодием в более позднем возрасте [5]. На протяжении менструального цикла клетки эндометрия удаляются путём апоптоза, что является ключевым феноменом предстоящего ремоделирования ткани, которое происходит в эндометрии при переходе от секреторной к пролиферативной фазе после менструации [6]. Колоссальную роль апоптоз играет в развитии и функционировании клеток иммунной системы. Так, например, во время развития Т-лимфоцитов апоптозу подвергаются те клетки, которые обладают нефункциональными или аутореактивными Т-клеточными рецепторами. Нарушение этого механизма элиминации иммунных клеток приводит к развитию иммунодефицитных и аутоиммунных состояний [7-9]. Развитие множества патологий ассоциировано именно с нарушением апоптоза и/или баланса между апоптозом и пролиферацией клеток. К таким патологиям относятся как редкие заболевания, например, синдактилия [10] и дефект развития нервной трубки [11], так и более распространённые, включая онкологические заболевания [12]. В норме в организме ежедневно за счёт апоптоза гибнут миллиарды клеток как вследствие физиологических процессов, описанных выше, так и вследствие воздействия различных стрессорных факторов, например, γ-излучения, действия химических соединений, теплового или холодового шоков и т.д. Поэтому исключительно важно избавляться от погибших клеток с целью поддержания тканевого гомеостаза и предотвращения высвобождения клеточного содержимого в окружающие ткани, поскольку это грозит развитием воспаления [13]. Клиренс апоптотических клеток, или эффероцитоз, осуществляют профессиональные (макрофаги, дендритные клетки) и непрофессиональные (эпителиоциты, эндотелиоциты, гепатоциты и др.) фагоциты. Эти клетки в данном контексте называют эффероцитами. Эффероцитоз является завершающей стадией апоптоза и необходим для поддержания гомеостаза и репарации тканей [14]. Доказана роль эффероцитоза в восстановлении тканей после повреждения. Так, например, при повреждении мышц провоспалительные моноциты проникают в повреждённую область и становятся провоспалительными макрофагами, которые стимулируют пролиферацию мышечных стволовых клеток. Эти провоспалительные макрофаги, эффероцитируя дебрис мышечных клеток, поляризуются в противовоспалительные макрофаги, которые стимулируют миогенез и образование новых функциональных миофибрилл [15]. При различных патологиях печени эффероцитоз также играет важную репаративную роль. Хроническое воспаление, ассоциированное с ожирением и приводящее к развитию неалкогольной жировой болезни печени [16], на ранних стадиях сдерживается именно за счёт эффероцитоза гепатоцитов, погибших вследствие липотоксичности, резидентными макрофагами, что поддерживает иммунный гомеостаз в печени и при снижении липидной нагрузки не позволяет развиваться заболеванию [17]. Zhang и соавт. предположили, что макрофаги используют полученные из эффероцитированных клеток метаболиты для противовоспалительного перепрограммирования, которое и приводит к восстановлению органов и тканей [18]. Похожий механизм был показан и для нейтрофилов, эффероцитирующих апоптотические тельца, сформировавшиеся из гепатоцитов после частичной гепатэктомии и попавшие в кровоток. Эффероцитоз нейтрофилами таких циркулирующих апоптотических телец приводил к их активации, однако без развития классических воспалительных реакций. Вместо этого эффероцитирующие нейтрофилы высвобождали различные факторы роста, включая фактор роста фибробластов-2 и фактор роста гепатоцитов, которые способствовали регенерации печени [19]. Помимо эндогенного репаративного механизма эффероцитоз также рассматривается в качестве одного из способов реализации регенеративных эффектов стволовых клеток, трансплантируемых при различных патологических состояниях [20]. Наиболее изученными в данном контексте стволовыми клетками, подвергающимися эффероцитозу после трансплантации, являются МСК. Достоверно доказано, что МСК при введении в организм реципиента просуществовали в нём не более 7 дней, и в последующем сигнал от них не детектировался, что связано с гибелью трансплантированных клеток по механизму апоптоза [21]. Несмотря на столь быструю гибель МСК, их трансплантация в большинстве случаев приводит к положительным терапевтическим эффектам [22]. Так же, как и другие апоптотические клетки, апоптотические МСК элиминируются при помощи эффероцитоза. Что особенно интересно, процесс клиренса апоптотических МСК влияет на фагоциты, осуществляющие этот процесс. Это влияние проявляется в изменении их фенотипа, секретома или дальнейшего поведения фагоцита. В совокупности это влияет на протекание и исход заболевания [23]. В данном обзоре рассмотрены базовые молекулярные механизмы эффероцитоза, клиренс апоптотических МСК и их терапевтические эффекты при различных патологиях в контексте их эффероцитоза различными типами фагоцитов. 1. МЕХАНИЗМ ЭФФЕРОЦИТОЗА Эффероцитоз (лат. efferro – захоронить) представляет собой частный случай фагоцитоза и направлен на фагоцитирование апоптотических клеток/телец без вовлечения иммунного ответа. Эффероцитоз можно разделить на несколько стадий: 1) распознавание эффероцитом апоптотической клетки; 2) взаимодействие эффероцита с апоптотической клеткой; 3) интернализация апоптотической клетки; 4) деградация апоптотической клетки внутри эффероцита. Схематически этапы эффероцитоза представлены на рисунке 1.
Распознавание апоптотической клетки эффероцитом происходит за счёт так называемых “find-me” сигналов – молекул, высвобождаемых клетками при апоптозе, включая нуклеотиды (ATP, UTP), липиды (сфингозин-1-фосфат, лизофосфатидилхолин) и хемокин фракталкин (CX3CL1). “Find-me” сигналы являются хемоаттрактантами для эффероцитов [14, 24]. После обнаружения апоптотической клетки эффероциту предстоит совершить выбор – фагоцитировать её или нет. На жизнеспособных клетках экспрессируются так называемые “don't eat me” сигналы (CD31, CD46, CD47) для предотвращения поглощения их фагоцитами; однако на апоптотических клетках происходит одновременная экспрессия “don't eat me” и “eat me” сигналов (к числу последних относятся фосфатидилсерин, кальрегулин) [25]. В связи с этим апоптотической клетке требуется избавиться или снизить экспрессию “don't eat me” сигналов для её успешного поглощения эффероцитом. CD47 является наиболее изученным и широко распространенным "don't eat me" сигналом, который экспрессируется практически на всех клетках организма. Этот трансмембранный гликопротеин, относящийся к семейству иммуноглобулинов, взаимодействует с иммунорецептором SIRPα, экспрессируемым на поверхности макрофагов, инициируя ингибирующие сигналы, которые предотвращают фагоцитоз макрофагами здоровых клеток. Индукция апоптоза приводила к снижению CD47 и CD31 на некоторых типах клеток, что активировало их эффероцитоз [26], однако полное отсутствие CD47 на клеточной мембране полностью предотвращало эффероцитоз [27]. Lv с соавт. показали, что не столько снижение экспрессии, сколько изменение локализации CD47 на плазматической мембране апоптотических клеток делает их доступными для эффероцитов [28]. Авторы продемонстрировали, что уровень CD47 на клеточной поверхности не снижался, но характер распределения CD47 изменялся во время апоптоза. В неапоптотических клетках молекулы CD47 группируются в липидных рафтах, тогда как в апоптотических клетках молекулы CD47 диффундируют по плазматической мембране вне рафтов. Кластеризация CD47 в липидных рафтах обеспечивает высокую авидность связывания с SIRPα на макрофагах, который также представлен в виде кластеров, что вызывает предотвращение фагоцитоза. Напротив, диспергирование CD47 на поверхности апоптотических клеток ассоциировано со значительным снижением авидности связывания с SIRPα и неспособностью запустить передачу ингибирующего сигнала. Таким образом, CD47 обычно кластеризуется в липидных рафтах на неапоптотических клетках, но диффундирует в плазматическую мембрану, когда происходит апоптоз; эта транслокация CD47 значительно снижает силу взаимодействия CD47-SIRPα, что приводит к фагоцитозу апоптотических клеток [28]. В настоящее время ось CD47-SIRPα рассматривается в качестве многообещающей терапевтической мишени в терапии широкого спектра заболеваний, ассоциированных с нарушением фагоцитоза/эффероцитоза, таких как неалкогольный стеатогепатит [29], сердечно-сосудистые [30], онкологические заболевания [31, 32], заболевания, ассоциированные со старением [33]. В случае снижения экспрессии и/или перераспределения в плазматической мембране "don't eat me" сигналов для эффероцита становятся доступными “eat-me” сигналы, которые позволяют ему прикрепиться к апоптотической клетке. Классическим “eat me” сигналом является фосфатидилсерин, транслоцирующийся с внутреннего на внешний монослой плазматической мембраны в процессе апоптоза за счёт механизма “flip-flop” [34], что делает его доступным для распознавания эффероцитом. Фосфатидилсерин на апоптотической клетке может распознаваться либо напрямую за счёт рецепторов, экспрессируемых на фагоцитах, таких как TIM (T-cell immunoglobulin and mucin domain receptor) (TIM-1, TIM-3, TIM-4) и SCARF-1 (scavenger receptor class F, member 1), либо через специфические растворимые молекулы-мостики, такие как белок S (PROS), Gas6 и MFG-E8, которые связываются одновременно с фосфатидилсерином на апоптотической клетке и рецепторными киназами ТАМ (Tyro3-Axl-Mertk) семейства, включая TYRO3, AXL и MerTK на фагоцитах. Например, MerTK связывается с фосфатидилсерином через такие молекулы-мостики, как Gas6 или белок S (PROS1) [35]. Кальретикулин (кальрегулин), ещё один “eat me” сигнал, в норме находится в эндоплазматическом ретикулуме, транслоцируется на поверхность апоптотической клетки и играет важную роль в апоптотическом клиренсе [36], что обусловлено его способностью связываться с белком CD91 (также известным как белок, связанный с рецептором белка низкой плотности, LRP-1), экспрессируемым на макрофагах, а также с компонентом комплемента C1q и маннозосвязывающим лектином (MBL). C1q и MBL – структурно и функционально схожие белки, члены семейства «защитного коллагена» — представляют собой молекулы распознавания образов (pattern recognition molecules), которые могут запускать быстрый усиленный фагоцитоз, что приводит к эффективному сдерживанию патогенов или удалению клеточного мусора, апоптотических клеток и иммунных комплексов [37]. Структурно оба белка представляют собой полипептиды, С-конец которых представлен глобулярным доменом, выполняющим функцию лектина, т.е. связывается с углеводами на поверхности патогенов и/или повреждённых и апоптотических клеток, тогда как N-конец представляет собой коллаген-подобный домен (повторяющиеся триплеты Gly-X-Y, где X часто представляет собой пролин, а Y – гидроксилизин или гидроксипролин) [37]. Таким образом, взаимодействуя с апоптотическими клетками, C1q и MBL действуют как опсонины [38]. После прикрепления C1q или MBL к апоптотической клетке кальретикулин, экспрессированный на её поверхности, связывается с коллагеновыми доменами обоих белков, а также с CD91 на поверхности макрофагов, способствуя вовлечению этого рецептора и стимулируя фагоцитоз. Эта серия событий инициирует макропиноцитоз, который приводит к поглощению апоптотической клетки [39]. Также “eat-me” сигналом служит снижение количества сиаловых кислот на мембране апоптотической клетки, которые в норме экранируют остатки сахаров гликокаликса, защищая от связывания их с лектиновыми рецепторами макрофагов [40]. Как только эффероцит прикрепился к апоптотической клетке, запускается процесс, направленный на реорганизацию актинового цитоскелета и последующее поглощение апоптотической клетки. Реорганизация актиновых филаментов направлена на формирование фагоцитарной чаши, которая за счёт выростов плазматической мембраны фагоцита окружает апоптотическую клетку и постепенно замыкается, формируя фагосому. Внутриклеточный сигналинг, управляющий формированием фагоцитарной чаши и разрастанием отростков вокруг апоптотической клетки, включает два потенциально избыточных, эволюционно консервативных пути, включающих LRP/GULP и Trio/RhoG/CrkII/Dock180/ELMO, которые могут действовать согласованно в регуляции активации Rac1 (белок из семейства Rho GTPаз), который является ключевым регулятором актинового цитоскелета в процессе интернализации апоптотической клетки [41]. После активации через любой из двух путей Rac1 индуцирует локализованную полимеризацию актина. Первый этап полимеризации актина известен как «нуклеация». На этом этапе формируется актиновое ядро, которое, по существу, представляет собой комплекс из трёх актиновых мономеров, из которого может удлиняться актиновая нить. Rac1 активирует факторы семейства WASP, SCAR и WAVE, которые рекрутируют комплекс ARP2/3 и функционируют вместе, чтобы создать ядро нуклеации актина. Помимо формирования ядра нуклеации для полимеризации актина de novo, комплекс ARP2/3 связывает актиновые нити, обеспечивая синтез нового актина, сохраняя при этом формирование сети и разветвление актина — процессы, которые необходимы для образования фагосом [14]. Выросты мембраны замыкаются над апоптотической клеткой и, таким образом, формируется фагосома. Фагосома, содержащая апоптотическую клетку, претерпевает созревание в цитоплазме эффероцита. Созревание фагосомы включает в себя несколько последовательных стадий, в конечном итоге приводящих к деградации апоптотической клетки: ранняя фагосома, поздняя фагосома и фаголизосома. Созревающая фагосома сливается с ранней эндосомой при помощи GTPазы Rab5 [42]. Rab5-содержащая ранняя фагосома рекрутирует ранний эндосомальный антиген 1 EEA1 (early endosome antigen 1), способствующий слиянию фагосомы с ранними эндосомами [43, 44]. При созревании фагосомы не формируется огромная везикула, т.к. происходит постоянное отпочковывание рециркулирующих эндосом от созревающей фагосомы. Основная роль ранних эндосом заключается в сортировке различных белков и липидов в компартменты рециркуляции и деградации. Следовательно, слияние фагосомы с ранней эндосомой необходимо для того, чтобы мембранные липиды и белки, участвовавшие в формировании фагосомы, смогли рециркулировать обратно в цитоплазматическую мембрану при помощи рециркулирующих малых эндосом, отпочковывающихся от ранней эндосомы. Кроме того, ранние эндосомы получают мембранные белки и гидролитические ферменты за счёт слияния секреторных везикул из сети транс-Гольджи, что важно для процесса созревания эндосом и, как следствие, слившихся с ними фагосом [45]. Rab5 также рекрутирует фосфатидилинозитол-3-киназу класса III (VPS34), катализирующую образование фосфатидилинозитол-3-фосфата [46]. Фосфатидилинозитол-3-фосфат способствует закреплению EEA1 на цитозольной поверхности созревающей фагосомы, что позволяет рекрутировать ещё одну GTPазу Rab7 [47]. Rab7 способствует слиянию фагосомы с поздней эндосомой, в это же время фагосома избавляется от Rab5. Этот переход осуществляется при помощи комплекса HOPS (vpsC-homotypic protein-sorting complex) [47]. GTPаза Rab7 – это маркер следующей стадии созревания фагосомы – стадии поздней фагосомы. GTPаза Rab7 необходима для рекрутирования RILP (Rab-interacting lysosomal protein), который связывается с динеин-динактиновым комплексом для ускоренного перемещения поздней фагосомы в цитоплазме [48]. На мембране накапливаются молекулы V-ATPазы (vacuolar ATPase), которые закачивают протоны H+ в фагосому и, тем самым, закисляют её [49]. Также на поздней фагосоме после слияния с поздней эндосомой появляются люминальные протеазы LAMP-1 и LAMP-2 (Lysosome-associated membrane proteins 1 и 2), которые способствуют слиянию поздней фагосомы с лизосомой, вследствие чего образуется фаголизосома [48, 50]. Фаголизосома содержит различные ферменты: протеазы, липазы, катепсины, гидролазы и NADPH-оксидазу, синтезирующую АФК (активные формы кислорода) [49]. V-ATPаза закисляет фагосому до оптимального значения рН, необходимого для работы протеолитических ферментов (pH 4.5–5.0), способствуя, тем самым, эффективной деградации апоптотических клеток. 2. БАЗОВЫЕ ХАРАКТЕРИСТИКИ МСК И ИХ ТЕРАПЕВТИЧЕСКИЙ ПОТЕНЦИАЛ МСК – фибробластоподобные негемопоэтические клетки, представляющие собой гетерогенную популяцию и соответствующие трём минимальным критериям: 1) адгезии к пластику при поддержании в стандартных условиях культивирования; 2) экспрессии поверхностных маркеров, включая CD73, CD90, CD105, и отсутствии экспрессии гемопоэтических маркеров, таких как CD45, CD34, CD14 или CD11b, CD79a или CD19 и HLA-DR; 3) способности дифференцироваться в клетки мезодермального ряда in vitro, остеобласты, адипоциты и хондробласты [51]. МСК присутствуют практически во всех тканях фетального и взрослого организмов и, соответственно, легко могут быть выделены из таких источников, как костный мозг, жировая ткань [52], периферическая [53] и менструальная [54] кровь, ткань эндометрия [55], пульпа зуба [56], экстраэмбриональные органы, включая пупочный канатик, пуповинную кровь, желе Вартона, ткани плаценты [57]. Уникальность МСК заключается в их обширном терапевтическом потенциале при различных патологических состояниях, доказанная в огромном количестве экспериментов на животных моделях заболеваний, а также в клинических испытаниях [58-60]. Примечательно, что не только сами МСК, но и их так называемые производные, включая экстраклеточные везикулы (экзосомы, апоптотические тельца), секретируемые растворимые факторы в составе кондиционированных сред, оказывают схожие терапевтические эффекты [20]. После трансплантации МСК и/или их производные стимулируют регенерацию повреждённых тканей, ангиогенез, оказывают антифибротическое и иммуномодулирующее действие. Терапевтическая эффективность МСК была показана для таких групп заболеваний, как сердечно-сосудистые [61, 62], нейродегенеративные [63], аутоиммунные [64, 65], различные патологии печени [66, 67] и желудочно-кишечного тракта [68] и др. Несмотря на столь глубокие и масштабные исследования, точные механизмы, лежащие в основе терапевтического действия МСК, остаются до конца не выясненными. Наиболее вероятным механизмом считается паракринный эффект этих клеток, связанный с секрецией широкого спектра цитокинов, хемокинов, ростовых факторов, а также с продукцией экстраклеточных везикул, загруженных различными РНК, белками, липидами, которые осуществляют полезную коммуникацию между клетками [69]. 3. АПОПТОЗ МСК IN VIVO Изначально считалось, что МСК оказывают терапевтические эффекты при введении в организм реципиента благодаря своему дифференцировочному потенциалу, т.е. замещая повреждённые/погибшие клетки. В ходе дальнейших исследований выяснилось, что МСК способны взаимодействовать с другими клетками и секретировать паракринные факторы, оказывающие влияние на повреждённую ткань. Однако в последующем стало очевидным, что МСК в организме реципиента персистируют непродолжительное время [70]. В итоге, исследователи пришли к выводу, что после трансплантации МСК подвергаются апоптозу независимо от способа их введения и степени их совместимости с реципиентом (ксеногенные, аллогенные или сингенные) [71]. Внутривенное введение – самый распространённый способ трансплантации МСК, в результате которого трансплантированные клетки "застревают" в капиллярах лёгких, где подвергаются апоптозу и элиминируются макрофагами в течение нескольких дней [21, 70]. Например, после внутривенного введения флуоресцентный сигнал от мышиных сингенных костномозговых МСК детектировался в течение 7 дней, так же, как и после введения в поджелудочную железу и подкожно, т.е. локально, причём МСК незначительно мигрировали из места их введения [21]. Несмотря на доказанность факта наличия апоптоза МСК после их трансплантации, относительно мало известно о физиологических индукторах и путях клеточной гибели трансплантированных клеток in vivo. При подкожном введении МСК формируют клеточные агрегаты, внутри которых создаются гипоксические условия и активируется HIF-1α-сигнальный путь. При этом, чем выше уровень гипоксии внутри такого клеточного агрегата, тем ниже жизнеспособность клеток [72]. Таким образом, гипоксия и дефицит питательных веществ могут являться одним из механизмов индукции апоптоза МСК после трансплантации, по крайней мере, в условиях локального введения [73, 74]. В индукции апоптоза трансплантированных МСК активное участие принимают различные типы иммунокомпетентных клеток реципиента. Например, в мышиной модели реакции трансплантат-против-хозяина (РТПХ) трансплантированные человеческие МСК подвергались апоптозу по МНС-независимому антиген-неспецифичному перфорин-гранзимовому механизму, который реализовывался за счёт активированных цитотоксических CD8+ T-лимфоцитов и CD56+ NK клеток. Не активированные Т- и NK клетки не индуцировали клеточную гибель МСК ни in vivo, ни in vitro [75]. В качестве наиболее очевидного механизма индукции апоптоза трансплантированных МСК чаще всего рассматривают классический Fas/FasL-путь. Это связано с тем, что МСК характеризуются высоким уровнем Fas (CD95) на своей поверхности; он служит рецептором соответствующего лиганда FasL, который в свою очередь экспрессируется иммунными клетками, включая Т-лимфоциты и NK клетки. В ранних работах было показано, что активированные Т-лимфоциты способны запускать апоптоз МСК именно по Fas/FasL-пути. В частности, на модели остеопороза было показано, что костномозговые МСК не дифференцируются в остеокласты из-за гибели, индуцированной активированными Т-лимфоцитами через Fas/FasL [76]. Fas-индуцированный апоптоз наблюдался в МСК после трансплантации в ишемизированном миокарде [77]. Здесь стоит отметить, что ишемия ткани сопровождается гипоксией; в таких условиях кардиомиоциты начинают секретировать высокие уровни FasL, а на поверхности МСК возрастает экспрессия Fas. Кроме того, гипоксия приводит к повышенной генерации АФК в МСК, что делает их более чувствительными к Fas-индуцированному апоптозу. В нормальных условиях, в отсутствие повышенной продукции АФК, МСК проявляли устойчивость к клеточной гибели, индуцированной FasL [77]. В целом, пока остаётся не выясненным вопрос, касающийся чувствительности/устойчивости МСК к апоптозу, индуцированному через Fas, поскольку в экспериментах in vitro были получены противоречивые данные. Например, Kennea с соавт. показали, что человеческие МСК, выделенные из фетальной крови, отвечают как на стауроспорин, так и на FasL дозозависимым увеличением апоптоза [78]. Стауроспорин – ингибитор протеинкиназ [79] – в данном случае использовался в качестве стрессового стимула, который, как известно, активирует митохондриальный путь клеточной гибели [79], а связывание Fas использовалось для проверки передачи сигнала через рецептор смерти. Эти результаты свидетельствуют о том, что в человеческих фетальных МСК могут быть активированы оба классических апоптотических пути [78]. С другой стороны, Götherström с соавт. продемонстрировали, что человеческие фетальные печёночные МСК были в большей степени чувствительны к TRAIL-индуцированному апоптозу, но не к апоптозу, индуцированному через Fas, по сравнению с человеческими взрослыми костномозговыми МСК [80]. Взрослые печёночные МСК также оказались устойчивы к Fas-индуцированному апоптозу в широком диапазоне концентраций FasL и анти-Fas антител [81]. В иммортализованных человеческих костномозговых МСК FasL индуцировал повышенную продукцию АФК, которые и служили стимулом к апоптозу. В присутствии антиоксиданта N-ацетил цистеина МСК не отвечали на FasL продукцией АФК и, следовательно, не подвергались апоптозу [82]. По некоторым данным, FasL в зависимости от дозы может оказывать плейотропные эффекты на МСК, не только индуцирует апоптоз, но стимулирует пролиферацию и поддерживает стволовое состояние [83]. Помимо CD95 (Fas) МСК экспрессируют, по меньшей мере, ещё два рецептора смерти DR4 и DR5, лигандом которых является TRAIL (TNF-related apoptosis-inducing ligand) [84, 85]. TRAIL может взаимодействовать с четырьмя мембраносвязанными рецепторами (DR4/TRAIL-R1, DR5/TRAIL-R2, DcR1/TRAIL-R3 и DcR2/TRAIL-R4), но только DR4 и DR5 содержат консервативный цитоплазматический регион, называемый доменом смерти (death domain (DD)), который необходим для проведения апоптотического сигнала после связывания TRAIL [86]. Szegezdi с соавт. показали, что сигнализация через DR4 и DR5 в человеческих костномозговых и пуповинных МСК была полностью блокирована, что делало их устойчивыми к TRAIL-индуцированному апоптозу даже при высоких дозах лиганда (500 нг/мл) [84]. С другой стороны, в МСК, выделенных из фетальной крови, полностью блокирован был только DR4, тогда как после связывания с DR5 апоптотическая сигнализация происходила [84]. Подобная устойчивость МСК к рецептор-опосредованному апоптозу может быть связана с высокой экспрессией в этих клетках антиапоптотических белков, включая c-FLIP, bcl-2, и низкой экспрессией проапоптотических белков, в частности, прокаспазы-8. Таким образом, до настоящего момента остаётся неизвестным, что же является триггером индукции апоптоза МСК после трансплантации. Тем не менее, очевиден тот факт, что гибель трансплантированных клеток является обязательным этапом последующих событий, приводящих в конечном итоге к терапевтическому эффекту. 4. ЭФФЕРОЦИТОЗ МСК ОКАЗЫВАЕТ ПРОТИВОВОСПАЛИТЕЛЬНЫЕ ЭФФЕКТЫ МСК, подвергшиеся апоптозу после трансплантации, эффероцитируются профессиональными (макрофаги, дендритные клетки) или непрофессиональными (эндотелиоциты, эпителиальные клетки, гепатоциты и другие соматические клетки) фагоцитами, которые являются клеточными посредниками в реализации терапевтических эффектов МСК. Например, в мышиной модели реакции трансплантат-против-хозяина (РТПХ) апоптотические МСК, введённые интраперитонеально, в основном эффероцитировались CD11b+ и CD11c+ фагоцитами в дренирующих брюшину лимфатических узлах, но не детектировались в лёгких и селезёнке. Напротив, при внутривенном введении в лёгких обнаружилось несколько популяций фагоцитов CD11bhighCD11cint, CD11bhighCD11c- и CD11b-CD11c+, которые поглощали апоптотические МСК [75]. Примечательно, что эффероцитированные апоптотические МСК приводили к повышению продукции индоламин-2,3-диоксигеназы (IDO, indoleamine-2,3-dioxygenase) фагоцитами после интраперитонеального, но не внутривенного введения, что вызывало выраженный иммуносупрессивный эффект в модели РТПХ [75]. IDO является катаболическим ферментом, участвующим в деградации незаменимой аминокислоты триптофана. Расщепляя ароматическое индольное кольцо триптофана, IDO инициирует выработку различных продуктов распада триптофана, называемых «кинуренинами», которые выполняют важные иммунорегуляторные функции [87]. Bikorimana с соавт. на мышиной модели РТПХ показали, что выраженный иммуносупрессивный эффект оказывала CD146high субпопуляция костномозговых МСК, но не CD146low клетки, трансплантированные интраперитонеально. Авторы предполагают, что этот эффект связан с лучшим эффероцитозом CD146high МСК CD11bhigh миелоидными клетками, и предлагают считать маркер CD146 “eat me” сигналом [88]. Ещё одним способом иммунорегуляции, ассоциированной с эффероцитозом апоптотических МСК, является поляризация эффероцитирующих макрофагов в противовоспалительный М2 фенотип [89]. M2 макрофаги характеризуются противовоспалительным профилем цитокиновой секреции. В частности, М2 макрофаги продуцируют высокие уровни IL-10 и TGF-β, и низкие уровни IL-12; для них также характерна секреция аргиназы-1 (Arg-1), которая снижает выработку NO за счёт ограничения биодоступности внутриклеточного L-аргинина, что приводит к ослаблению воспалительного повреждения тканей, и хемокинов CCL17, CCL24 и др. [90, 91]. Благодаря такому профилю секреции, M2 макрофаги способствуют регенерации ткани и заживлению ран, обладают проангиогенным свойством за счёт секреции фактора роста сосудистого эндотелия (VEGF), а также высокой способностью к фагоцитозу [89]. Апоптотические МСК, индуцируя М2 поляризацию макрофагов, т.е. переключение провоспалительного М1 фенотипа в противовоспалительный М2 фенотип, также стимулировали повышенную экспрессию в макрофагах Arg-1 и MerTK (Mer-tyrosine kinase), что способствовало снижению аллостимуляторной активности таких М2 макрофагов в смешанной культуре с Т-лимфоцитами [92], что говорит о способности макрофагов модулировать адаптивный Т-клеточный ответ. Поляризации макрофагов в М2 фенотип способствует сфингозин-1-фосфат (S1P), высвобождаемый апоптотическими клетками, который взаимодействует с рецептором S1PR1 на макрофагах, а также выполняет функцию хемоаттрактанта для регенеративных/ранозаживляющих макрофагов, способствуя их миграции в очаг воспаления [93, 94]. S1P запускает эритропоэтин-сигнальный путь, приводящий к повышенной экспрессии MerTK на макрофагах, что способствует клиренсу апоптотических клеток [95]. Моноциты, выделенные из периферической крови человека, эффективно поглощали апоптотические МСК, которые были получены путём индукции Fas-зависимого апоптоза человеческих пуповинных МСК. Это приводило к появлению иммуносупрессорного фенотипа. После фагоцитоза апоптотических МСК человеческие классические CD14++CD16- моноциты поляризовались в неклассический CD14++CD16+CD206+ фенотип и начинали экспрессировать PD-L1 (programmed death ligand-1) и IL-10, тогда как экспрессия TNF-α существенно снижалась. Такие моноциты, эффероцитировавшие апоптотические МСК, индуцировали формирование Foxp3+ регуляторных T-клеток in vitro [96]. Кроме того, моноциты, поглотившие апоптотические МСК, вызывали подавление пролиферации Т-лимфоцитов, активированных через CD3/CD28, за счёт повышенной секреции IDO, COX2 (циклооксигеназа 2) и PGE2 (простагландин Е2) [97]. У пациентов с тяжёлой стероидорезистентной РТПХ, которые отвечали на терапию МСК уровень PGE2 существенно повышался [97]. Фосфатидилсерин, локализованный на внешнем монослое плазматической мембраны апоптотических МСК, опосредует иммуносупрессорное действие на активированные CD4+ Т-лимфоциты [98]. Так, апоптотические тельца, полученные путём обработки стауроспорином мышиных костномозговых МСК, напрямую взаимодействовали с активированными CD4+ Т-лимфоцитами за счёт связывания фосфатидилсерина, экспонированного на поверхности апоптотического тельца, и TIM-3, экспрессируемого на Т-клетке [98]. Это взаимодействие приводило к ингибированию внутриклеточной трансдукции сигнала через Т-клеточный рецептор в Т-лимфоцитах без поглощения апоптотических МСК, как происходило в случае с макрофагами. Примечательно, что апоптотические МСК оказывали иммуносупрессорное действие только на активированные эффекторные CD4+ Т-клетки, и не оказывали эффектов на не активированные лимфоциты. Апоптотические МСК дозо-зависимо ограничивали экспансию эффекторных популяций, включая Th1 (IFNγ+CD4+), Th17 (IL-17A+CD4+) и Th2 (IL-4+CD4+), что сопровождалось снижением уровней цитокинов IFNγ, IL-17A и IL-4, in vitro и in vivo и оказывали поддерживающий эффект на регуляторные Foxp3+CD4+ T-клетки [98]. Схематически терапевтические эффекты апоптотических МСК представлены на рисунке 2. Таким образом, апоптотические МСК могут осуществлять иммуномодулирующую функцию как после эффероцитоза моноцитами/макрофагами, приобретающими иммуносупрессорный фенотип, так и за счёт прямого взаимодействия с эффекторными Т-клетками.
5. АПОПТОТИЧЕСКИЕ МСК СПОСОБСТВУЮТ РЕГЕНЕРАЦИИ ТКАНИ И ЗАЖИВЛЕНИЮ КОЖНЫХ РАН Регенеративный потенциал апоптотических МСК ассоциирован не только с иммуномодуляцией, но и с воздействием на тканеспецифичные клетки, что приводило к усилению ангиогенеза, снижению фиброза, стимуляции резидентных стволовых клеток и другим терапевтическим эффектам [89, 99-101]. На мышиной модели заживления раны было показано, что введение сингенных апоптотических костномозговых МСК, полученных путём обработки стауроспорином, в место вырезанного лоскута кожи приводило к меньшей инфильтрации воспалительных клеток, меньшей площади рубцов и более интегрированной структуре кожи с вновь сформированным эпителием, кроме того отмечено восстановление некоторых волосяных фолликулов, а отложения коллагена были более организованными по сравнению с контрольными животными [89]. Подобный терапевтический эффект апоптотических МСК был связан с их эффероцитозом F4/80 макрофагами и их последующей поляризацией в CD206+ М2 фенотип, который секретирует высокие уровни противовоспалительных цитокинов IL-10 и TGF-β [89]. Помимо этого, М2 макрофаги, эффероцитировавшие апоптотические МСК, эффективно рекрутировали в место повреждения кожные фибробласты, усиливая их миграцию, что в свою очередь способствовало заживлению раны [89]. Ma с соавт. показали, что внутрикожно введенные апоптотические МСК поглощались кожными МСК и МСК волосяных фолликулов, активируя в них Wnt/β-катенин сигнальный путь и стимулируя регенеративный потенциал [99]. Эти же авторы продемонстрировали на мышиной модели заживления раны, что апоптотические МСК, введённые внутривенно, стимулировали более быстрое восстановление кожного повреждения, а также усиливали рост волос, в том числе за счёт стимуляции пролиферации соответствующих резидентных МСК [99] (рис. 2). На крысиной модели инфаркта миокарда было показано, что сингенные костномозговые МСК, введённые локально в зону инфаркта, быстро подвергались апоптозу, генерируя большое количество апоптотических телец, которые поглощались преимущественно PECAM1/CD31-позитивными эндотелиальными клетками в пограничной с инфарктом зоне [100]. В экспериментах in vitro было продемонстрировано, что апоптотические МСК, поглощённые эндотелиоцитами, усиливали их миграцию, повышали пролиферацию и стимулировали ангиогенез за счёт увеличения экспрессии проангиогенных генов (ANGPT1 и KDR) и уменьшения антиангиогенных генов (THBS1 и VASH1) [100]. Механизм стимуляции ангиогенеза апоптотическими МСК был ассоциирован с повышенным биогенезом лизосом и аутофагии за счёт увеличения экспрессии TFEB (транскрипционный фактор ЕВ) [100]. TFEB — мастер-ген, регулирующий лизосомы и аутофагию, активно участвующий в процессе ангиогенеза и постишемической регенерации тканей [102]. Таким образом, в основе восстановления сердечной мышцы в данном исследовании лежит стимуляция ангиогенеза апоптотическими МСК через регуляцию аутофагии в реципиентных эндотелиоцитах. Такой же механизм стимуляции ангиогенеза, опосредованный через TFEB и аутофагию, был показан на модели регенерации пульпы зуба [101]. Апоптотические МСК пульпы зуба эффероцитировались эндотелиоцитами, стимулируя в них повышение экспрессии проангиогенных белков и белков, участвующих в клеточной миграции (HIF-1α, VEGF, ANG2 и MMP2), что приводило к реваскуляризации ткани и регенерации пульпы [101] (рис. 2). На мышиной модели пародонтита было показано, что сингенные апоптотические костномозговые МСК, полученные за счёт гипоксии, значительно ингибировали дифференцировку остеокластов и резорбцию альвеолярной кости [73]. Оказалось, что эти апоптотические МСК поглощались преостеокластами и доставляли в них miR-223-3p, которая оказывала значительное ингибирующее действие на остеокластогенез, что в совокупности приводило к уменьшению деструкции кости при пародонтите [73] (рис. 2). На двух моделях апоптоз-дефицитных мышей (MRL/lpr и Casp3−/−) было показано, что резидентные костномозговые МСК утрачивали способность дифференцироваться в остеоциты, вследствие чего развивалась остеопения [103]. Аллогенные апоптотические МСК, введённые внутривенно таким животным, эффективно поглощались резидентными костномозговыми МСК, восстанавливая их дифференцировочный потенциал в клетки мезодермального ряда за счёт активации Wnt/β-катенин сигнального пути, что способствовало снижению признаков остеопении. Такие же терапевтические эффекты экзогенно введенных апоптотических МСК наблюдались при остеопении у мышей с овариэктомией [103] (рис. 2). На мышиной модели сахарного диабета 2 типа было продемонстрировано, что внутривенно введённые апоптотические МСК в течение 24 ч накапливались в печени и эффективно поглощались как резидентными печёночными макрофагами, клетками Купфера, так и рекрутированными в печень моноцитами [104]. Эффероцитоз апоптотических МСК приводил к функциональному регулированию гомеостаза печёночных макрофагов, что проявлялось в виде снижения активации клеток Купфера, поляризации макрофагов в противовоспалительный М2 фенотип и значительного уменьшения рекрутинга и инфильтрации моноцитов и других иммунных клеток, поддерживающих воспаление [104]. В совокупности эти события способствовали восстановлению ткани печени, улучшению непереносимости глюкозы и снижению инсулинорезистентности [104]. Внутривенно введенные костномозговые МСК, подвергшиеся апоптозу в печени после трансплантации, эффективно модулировали баланс провоспалительных Ly6Chigh и противовоспалительных макрофагов Ly6Clow в мышиной модели CCl4-индуцированного фиброза. Такая модуляция и восстановление гомеостаза печёночных макрофагов была связана с несколькими механизмами: апоптотические МСК вызывали прямое переключение провоспалительных макрофагов Ly6Chigh в противовоспалительные Ly6Clow и предотвращали рекрутинг в повреждённую печень Ly6Chigh моноцитов из кровотока, тем самым существенно снижая соотношение Ly6Chigh/Ly6Clow в печени. Ly6Clow макрофаги, эффероцитировавшие апоптотические МСК, начинали продуцировать высокие уровни матриксной металлопротеиназы 12 (ММР12), которая участвует в деградации внеклеточного матрикса, что приводило к разрешению фиброза [105] (рис. 2). Исходя из вышеописанных примеров, можно предположить, что эффероцитоз трансплантированных МСК как профессиональными, так и непрофессиональными фагоцитами может являться обязательным и неотъемлемым этапом сложного механизма их терапевтического действия. ЗАКЛЮЧЕНИЕ Апоптоз трансплантированных МСК и их последующее поглощение эффероцитами изменяет свойства последних. Этот эффект распространяется на резидентные тканеспецифичные эффероциты, на эффероциты, рекрутированные из кровотока, а также на некоторые собственные стволовые клетки реципиента, в том числе МСК. У таких эффероцитов меняется фенотип и секретом, что приводит к кардинальному изменению их функциональной активности и, в свою очередь, оказывает влияние на течение и исход заболеваний, которыми страдает реципиент. Показано, что в большинстве случаев основными эффероцитами апоптотических МСК выступают макрофаги, что, вероятно, и обеспечивает проявление многих иммуномодулирующих эффектов МСК. Кроме того, в ряде работ, показано, что эффероцитами апоптотических МСК могут выступать и не иммунные клетки, такие как эндотелиоциты или резидентные стволовые клетки. В первом случае это приводит к стимуляции ангиогенеза, тогда как в случае эффероцитоза резидентными стволовыми клетками активируются регенеративные процессы в тканях, направленные на восстановление повреждённых клеток. Несмотря на публикации, демонстрирующие положительные терапевтические эффекты, связанные с эффероцитозом МСК, эта тема остаётся ещё недостаточно изученной. Однако она выглядит весьма перспективной как с точки зрения выяснения механизмов терапевтического действия МСК, так и с точки зрения разработки нового класса препаратов для терапии широкого спектра заболеваний. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов. БЛАГОДАРНОСТИ Авторы выражают благодарность графическому дизайнеру Кристине Александровне Лазуткиной за помощь в подготовке иллюстраций. ФИНАНСИРОВАНИЕ Исследование выполнено за счёт гранта Российского научного фонда №23-15-00149. КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|