|
Молекулярный профиль опухолевой клеточной линии HepG2
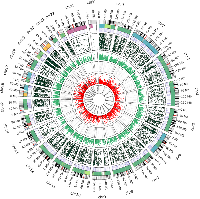
Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича, Ключевые слова: HepG2; клеточная линия; омикс данные; регуляция генов; молекулярный портрет DOI: 10.18097/BMCRM00239 ВВЕДЕНИЕ За последние десятилетия молекулярные биологи значительно расширили знания об "омах": геноме, метиломе, транскриптоме, протеоме и метаболоме для различных организмов и клеточных линий. Однако, несмотря на накопленные данные, взаимосвязи между уровнями реализации генетической информации остаются фрагментарными [1]. Пробелы в понимании взаимосвязей ограничивают возможности интерпретации наблюдаемых эффектов и могут приводить к некорректной постановке экспериментов, что усложняет поиск и разработку новых биомаркеров и методов лечения заболеваний. Клеточные линии часто используют как унифицированный и доступный объект исследований, поскольку они обеспечивают воспроизводимый молекулярный профиль и уменьшение количества повторностей в биомедицинских исследованиях [2, 3]. Убеждённость в стабильном молекулярном портрете клеточных линий до 15-20 пассажа (в зависимости от типа клеток) ослабило исследование одной из популярных клеточных линий HeLa [4]. Группа под руководством Liu проиллюстрировала наличие трёх подтипов клеток с различными геномными и транскриптомными профилями. Это подчёркивает важность изучения гетерогенности клеточных линий на разных молекулярных уровнях [4]. Особое внимание заслуживает опухолевая линия печени HepG2, частично сохранившая свойства гепатоцитов. Данную культуру клеток часто используют в качестве модели для токсикологических и фармакологических исследований, а также для изучения рака. На 2024 год в базе данных PubMed по запросу "HepG2" находится 40059 статей, что делает её четвертой по популярности клеточной линией после HeLa (рак шейки матки), MCF-7 (рак молочной железы) и A549 (рак лёгкого). Мы собрали молекулярный портрет опухолевой клеточной линии HepG2 на основе молекулярно-генетических данных на геномном, метиломном, транскриптомном, эпитранскриптомном и протеомном уровнях, что в итоге позволит более корректно использовать данную линию в экспериментах и минимизировать ошибки при интерпретации результатов. МАТЕРИАЛЫ И МЕТОДЫ Данные Для анализа клеточной линии HepG2 были использованы как собственные, так и общедоступные данные. Собственные данные включали результаты цитогенетического анализа, а также полногеномного (WGS), метиломного (WGBS), транскриптомного (RNA-seq), транслятомного (Polysome-seq) и протеомного (LC-MS/MS) профилирований, ранее полученные для одного образца [5]. Необработанные собственные данные WGS, WGBS, RNA-seq и Polysome-seq были загружены в NCBI SRA: PRJNA765908, PRJNA956723 и PRJNA972889, а протеомные данные доступны в Mendeley Data [6]. Для геномного и транскриптомного секвенирований использовали данные из NCBI SRA, полученные с помощью технологии Illumina. Критерии для поиска полногеномных и эпигеномных данных: >100 млн прочтений, длина чтений 100 пар нуклеотидов (п.н.), метод WGS/WGBS, для транскриптомных данных: >20 млн прочтений, длина чтений >60 п.н. Суммарно найдено восемь образцов полногеномного секвенирования HepG2, доступные эпигеномные данные не соответствовали критериям. В соответствии с критериями найдено 40 транскриптомных образцов HepG2. В PRIDE найдено 11 протеомных данных HepG2, согласно следующим критериям: данные должны быть представлены «сырыми» файлами и получены из необработанных клеток. Дополнительно был проведён поиск статей с опубликованными данными цитогенетического анализа для клеточной линии HepG2. В результате было найдено шесть результатов кариотипирования. Цитогенетический анализ Цитогенетическое исследование проводили с помощью G-дифференциального окрашивания 20 метафазных пластинок для определения хромосомного состава клеток. Анализ данных секвенирования нового поколения Для анализа собственных и опубликованных данных использовали одинаковые протоколы. Данные WGS, WGBS и RNA-seq (Illumina) были представлены в виде необработанных файлов fastq. Первый этап анализа включал оценку качества с использованием программы FastQ. Вторым этапом чтения низкого качества и адаптеры удаляли с помощью программы TrimGalore. Данные транскриптомного и транслятомного профилирований (Oxford Nanopore) были представлены в формате fast5. Первый этап обработки включал распознавание азотистых оснований с помощью программы Guppy, результатом которого были файлы fastq. Затем файлы выравнивались с помощью алгоритма Minimap2. Полногеномное секвенирование. Файлы fastq после фильтрации выравнивали на референсный геном человека (Ensembl GRCh38) с использованием алгоритма Bowtie2. Полученные bam-файлы использовали для оценки копийности хромосом с помощью программы mosdepth. Для детекции несинонимичных однонуклеотидных замен (SNP) и инделов использовались рекомендации GATK. Эпигеном. Идентификацию CpG участков выполняли с помощью алгоритма Bismark с выравниванием на геном человека (Ensembl GRCh38). Транскриптомное профилирование. Анализ экспрессии генов и изоформ, полученных с помощью Illumina и ONT, был выполнен программой Salmon. Экспрессию транскриптов рассчитывали в TPM (транскрипты на миллион), а экспрессию генов — суммированием TPM соответствующих транскриптов. Эпитранскриптом. Анализ данных прямого секвенирования РНК (direct RNA-seq) клеточной линии HepG2 для поиска сайтов модификации N6-метиладенозина (m6A) проводили с использованием программы Nanopolish и алгоритма m6Anet. Участки с коэффициентом модификации менее 0.9 исключали из анализа. Транслятомное профилирование. Данные транслятомного профилирования в двух повторах были получены с помощью Oxford Nanopore и анализировались по тому же протоколу, что и транскриптомные данные прямого секвенирования РНК. Протеомное профилирование. Протеомные данные для клеточной линии HepG2 были проанализированы по единому протоколу с учётом рекомендации протеомного сообщества [7]. Данные преобразовывали в формат mgf с помощью программы MSConvert и анализировали в программе SearchGUI с использованием алгоритма X!Tandem. Для полуколичественного анализа использовался индекс нормированных спектральных коэффициентов обилия (NSAF). Референсная база данных включала канонические и образующиеся в результате альтернативного сплайсинга последовательности белков для человека из SwissProt. Опубликованные протеомные данные также анализировали в SearchGUI с уникальными параметрами для каждого образца. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Анализ кариотипа Клеточная линия HepG2 характеризуется гипердиплоидным кариотипом, содержащим в среднем от 50 до 60 хромосом [8–11]. Анализ собственного и опубликованных кариотипирований клеток HepG2 выявил устойчивые хромосомные аномалии, включая транслокацию между короткими плечами хромосом 1p и 21p, тетрасомию хромосомы 20 и трисомию хромосомы 6 с потерей её короткого плеча. Также наблюдались и отличия для хромосом 16 и 17 между собственными и опубликованными данными [10]. Мы провели анализ глубины покрытия и GC состава для всех 10 образцов полногеномного секвенирования клеточной линии HepG2. На основе этих данных была получена анеуплоидная карта с высоким разрешением, выявившая значительные изменения числа копий в хромосомных областях клеточной линии HepG2. В 70% образцов обнаружено пять кластеров копийности. В среднем, геном клеточной линии HepG2 имеет 65% базового числа копий (CN=2). Результаты кариотипирования клеток HepG2 подтверждены анализом покрытия хромосом на геномном уровне. Сравнение с опубликованными данными показало стабильные хромосомные аномалии: транслокация между короткими плечами хромосом 1p и 21p, трисомия хромосомы 2, частичная трисомия хромосомы 6 (q1-q21), делеция короткого плеча хромосомы 9, тетрасомия хромосомы 20, потеря длинного плеча Y хромосомы и делеция коротких плеч у хромосом, имеющих спутники (SAT-хромосомы) – 13, 14, 15, 21 и 22. Таким образом, кариотип клеточной линии HepG2 сохраняет стабильные ключевые изменения, подтверждённые как собственными исследованиями, так и опубликованными данными, несмотря на различия межлабораторной воспроизводимости. Анализ генетических вариантов Сопоставление собственных и опубликованных данных на уровне несинонимичных однонуклеотидных замен показало, что только 7% вариантов встречались во всех образцах, 5,3% — в половине образцов, и 53% — только в одном или двух образцах. Наибольшее количество мутаций обнаружено в генах семейства MUC (муцины) и HLA (антиген человеческого лейкоцита). Муцины участвуют в формировании защитных слизистых барьеров, и их мутации могут способствовать прогрессированию опухоли и метастазированию. Гены HLA играют ключевую роль в иммунном ответе, и их мутации связаны с уклонением опухоли от иммунного надзора [12]. Такие значительные геномные аберрации подчеркивают важность генетического мониторинга для обеспечения надёжности и воспроизводимости результатов исследований. В результате анализ полногеномных данных также было выявлено отсутствие 1216 белок-кодирующих генов (БГК). Анализ метиломных данных Анализ метиломных данных для клеточной линии HepG2 показал, что 44% генома подверглось метилированию, что соответствует общему уровню метилирования в клетках человека [13]. Было определено 23637476 сайтов метилирования в 17369 БКГ. Анализ транскриптомных данных В результате анализа транскриптомных данных клеточной линии HepG2, полученных с помощью технологии Illumina, в среднем была обнаружена экспрессия 12602 генов и 32583 транскриптов при TPM >0.1. Коэффициент корреляции Спирмена между образцами составил 0.82 на уровне транскриптов и 0.93 на уровне генов. Для оценки гетерогенности клеток HepG2 были сопоставлены опубликованные данные транскриптомного профилирования 42 образцов. Между образцами выявлена сильная положительная корреляция: по данным экспрессии генов коэффициент корреляции Спирмена составил 0.88, а по данным экспрессии транскриптов — 0.77. Примечательно, что, несмотря на высокие значения корреляции, образцы разделились на два кластера. В первом кластере преобладали данные из китайских институтов, тогда как во втором — из институтов США, Японии, Южной Кореи, Турции, Колумбии, Италии и России. Анализ дифференциальной экспрессии выявил 2824 гена с различной экспрессией между этими двумя кластерами. Между кластерами была отмечена дисрегуляция генов, участвующих в метаболизме лекарств и детоксикации. Также были выявлены различия в путях "Окисление с помощью цитохрома P450" и "Метаболизм эстрогенов". Снижение экспрессии этих генов указывает на потенциальное ухудшение способности клеток к метаболизму и детоксикации лекарств, что может влиять на точность и значимость анализов метаболизма лекарств, проводимых с использованием клеток HepG2 [14]. Анализ эпитранскриптомных данных В клеточной линии HepG2 в пяти технических повторах суммарно обнаружено 3968 возможных сайтов m6A модификаций в 1396 генах. В результате сравнения была показана низкая воспроизводимость: только 6.3% (250 из 3968) возможных сайтов m6A были обнаружены во всех повторностях. Однако все гены с выявленными сайтами m6A модификаций демонстрировали ассоциированность с процессами убиквитилирования, несмотря на низкую воспроизводимость [15]. Таким образом, при наличии множественных технических повторов возможно использовать совокупную информацию для повышения надёжности данных. Анализ транслятомных данных За последние десять лет значительно увеличилось понимание роли трансляции как ключевого регулятора экспрессии генов. Транслятом выступает промежуточным звеном между транскриптомом и протеомом. Анализ транслятомного профилирования собственного образца клеточной линии HepG2 показал экспрессию 10461 гена и 22513 транскриптов при TPM >0.1. Анализ протеомных данных По результатам анализа нашего образца в трёх повторах было выявлено 1027 белков (NSAF >0). В результате анализа протеомного профилирования опубликованных данных для 14 образцов клеточной линии HepG2 были получены низкие значения корреляции Спирмена между данными полуколичественной оценки NSAF – 0.46. ЗАКЛЮЧЕНИЕ В ходе исследования установлено, что клеточная линия HepG2 демонстрирует в целом высокую степень стабильности на геномном и транскриптом уровнях, однако образцы из Китая требуют более пристального внимания при переносе результатов транскриптомных и протеомных экспериментов. В частности для клеток HepG2, выявлены устойчивые хромосомные перестройки, такие как транслокация между короткими плечами хромосом 1p и 21p, тетрасомия хромосомы 20, потеря короткого плеча у всех SAT-хромосом и длинного плеча Y хромосомы. При отсутствии на геномном уровне 1216 БКГ, на транскриптомном уровне экспрессируется 12602 гена, из которых только 10461 детектируются на уровне транслятов, а на протеомном уровне идентифицировано лишь 1027 генов (рис. 1).
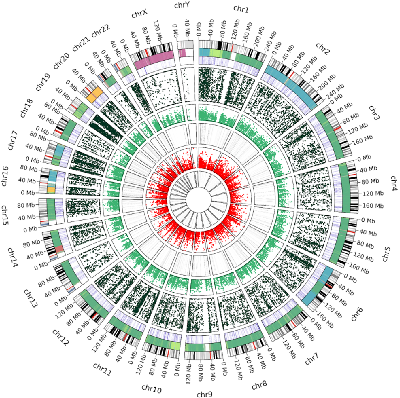
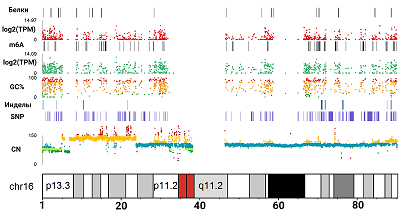
Впервые была построена подробная молекулярная карта клеточной линии HepG2 для каждой хромосомы (рис. 2), позволяющая оценить все молекулярные события на каждом участке генома. В качестве примера такой интеграции на рисунке 3 приведена хромосома 16 – благодаря полногеномному анализу уточнено, что не вся хромосома находится в трисомии, как следует из цитогенетического анализа. В середине короткого плеча (p13.2-12.2), находящегося в тетрасомном состоянии, уровень метилирования, экспрессия транскриптов и транслятов ниже, чем в целом для остальных участков хромосом, хотя количество сайтов m6A чуть выше. Данный регион содержит 122 БКГ, включая 10 из 24 генов, относящихся к семейству белков, взаимодействующих с ядерным поровым комплексом (Nuclear pore complex-interacting protein family member). Среди этих 122 генов 82 были детектированы на транскриптомном уровне, 78 на транслятомном и 6 на протеомном.
Таким образом, полученные результаты способствуют как более качественной интерпретации получаемых данных для клеточной линий HepG2, так и дальнейшей расшифровке взаимосвязей регуляции клеточных процессов. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Работа не содержит каких-либо исследований с участием людей или использованием животных в качестве объектов. БЛАГОДАРНОСТИ Эксперименты выполнены с использованием оборудования ЦКП “Протеом человека” Научно-исследовательского института биомедицинской химии им. В.Н. Ореховича. Кариотипирование выполнялось в ЦКП «Группа геномных технологий» в Институте биологии развития им. Н.К. Кольцова РАН. ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021-2030 годы) (№122030100168-2). КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|