|
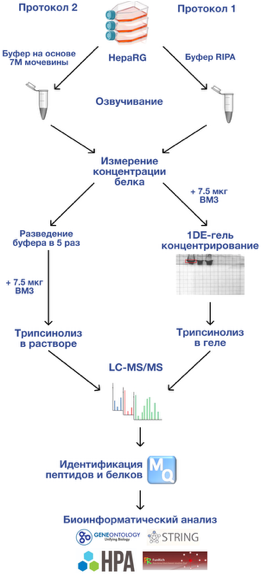
ВАЛИДАЦИЯ ПРОТОКОЛА ПРОБОПОДГОТОВКИ НА ОСНОВЕ 1-DE-ГЕЛЬ КОНЦЕНТРИРОВАНИЯ ДЛЯ ПРОТЕОМНОГО АНАЛИЗА КЛЕТОЧНОЙ КУЛЬТУРЫ HEPARG Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121, Москва, Погодинская ул., 10; *e-mail: avbolochenkov75@gmail.com Ключевые слова: HepaRG; ВМ-3 (Bifunctional cytochrome P450/NADPH-P450 reductase, cyp102A1); 1DE-гель концентрирование; LC-MS/MS; IdentiProt; белки метаболизма DOI: 10.18097/BMCRM00290 ВВЕДЕНИЕ Первичные гепатоциты человека признаны стандартом для изучения функций печени и метаболизма лекарственных средств (ЛС) [1]. Они активно применяются в научных исследованиях и оценке безопасности препаратов. Однако использование первичных гепатоцитов имеет ряд ограничений: клетки, полученные от разных доноров, сильно различаются между собой, быстро теряют функциональность вне организма и неспособны бесконечно делиться in vitro [2]. Большинство существующих клеточных линий печени человека не подходят в полной мере для замены первичных гепатоцитов. Примером служит линия HepG2, которая, будучи самой популярной лабораторной моделью, демонстрирует низкий уровень экспрессии генов, отвечающих за метаболизм ЛС [3]. Сегодня большое внимание привлекает клеточная линия HepaRG — уникальная бипотентная клеточная линия гепатомы человека, способная под действием диметилсульфоксида (ДМСО) дифференцироваться в два различных типа клеток: зрелые гепатоциты и гепатоцилиарные клетки [4]. Клетки HepaRG заняли лидирующие позиции среди всех иммортализованных линий клеток печени человека, которые были разработаны на сегодняшний день, в частности, для исследований гепатотоксичности [5]. Клетки данной линии производит ферменты первого (цитохромы Р450), второго (UDP-глюкоронозилтрансфераза) этапа метаболизма ксенобиотиков, а также регуляторные и специализированные белки печени (гаптоглобин, альдолаза B, альфа-фетопротеин, тиоредоксин) и ферменты антиоксидантной защиты (глутатионсинтазу, гамма-глутамилцистеинсинтазу, глутатионредуктазу) [6]. Это свойство позволяет применять клеточную линию HepaRG для оценки воздействия ЛС и исследований метаболизма ксенобиотиков, превосходя другие известные клеточные линии, например, HepG2. Протеомное профилирование клеточной линии HepaRG актуально для учёных и специалистов фарминдустрии, так как помогает в разработке новых терапевтических подходов, идентификации мишеней лекарств, поиска биомаркеров и оценки эффективности терапии. Этот метод позволяет одновременно изучать тысячи белков, их взаимодействие с лекарствами и изменения, происходящие в организме в ответ на лечение. Процесс пробоподготовки — важнейший этап любого протеомного анализа. Для разрушения клеток применяют механический лизис, экстракцию проводят с использованием детергентов [7]. Выбор подходящего детергента для извлечения белков является сложной задачей, так как многие из них могут ингибировать трипсин в ходе пробоподготовки, подавлять ионизацию пептидов и генерировать большое количество заряженных молекул, которые мешают масс-спектрометрическому анализу и увеличивают количество шумов [8]. Для солюбилизации белков используют ионные детергенты, такие как додецилсульфат натрия (ДСН), дезоксихалат натрия (ДОХ), неионные (например, Тритон X-100, NP-40, CHAPS), а также хаотропные агенты (мочевина или тиомочевина). Затем белки подвергают восстановлению и алкилированию остатков цистеина, что стабилизирует структуры и препятствует образованию дисульфидных мостиков. Ферментативный гидролиз белков обычно выполняют трипсином, который предпочтительно разрезает белки вблизи лизина и аргинина, облегчая последующее выделение и идентификацию пептидов [9]. Существует два варианта трипсинолиза: в растворе и в геле. Последний вариант обеспечивает лучшее очищение от примесей, хотя и осложняется увеличением числа образцов и трудоемкостью процесса. Правильный выбор стратегии пробоподготовки оказывает решающее влияние на успех протеомного анализа. ДСН часто используют для лизиса клеток и растворения мембранных белков при протеомном анализе различных тканей и клеток человека. Однако ДСН препятствует трипсинолизу и последующему масс-спектрометрическому анализу, поэтому его необходимо удалять после солюбилизации. Одним из лучших способов удаления ДСН является электрофорез в полиакриламидном геле (ДСН-ПААГ). Однако процесс разделения белков в ПААГ генерирует большое количество образцов, что увеличивает стоимость и сложность эксперимента. Помимо этого, возможны потери пептидов во время промывания геля и экстракции. Недавно была предложена методика 1DE-гель концентрирования, которая представляет собой усечённый вариант ДСН-ПААГ, при котором белки не разделяются, а концентрируется перед входом в разделяющий гель [10]. Полоса геля, содержащая все белки, вырезается и подвергается трипсинолизу для последующего масс-спектрометрического анализа. Такой способ пробоподготовки позволяет сократить количество проб до одной общей фракции, существенно упростив процедуру и увеличив воспроизводимость результатов. Целью данной работы было сравнить два протокола пробоподготовки: 1) на основе 1DE-гель концентрирования и трипсинолиза в растворе; 2) на основе трипсинолиза в растворе для анализа протеома клеточной линии HepaRG с применением технологий жидкостной хроматографии и тандемной масс-спектрометрии (LC-MS/MS). МЕТОДИКА В работе были использованы следующие реактивы: Трис(гидроксиметил)аминутометан (Трис), бикарбонат аммония (NH4HCO3), ацетонитрил (ACN, «Sigma-Aldrich», США), йодацетамид (IAA, «Sigma-Aldrich»), натрия хлорид (NaCl), натрия карбонат (Na2CO3), натрия гидрокарбонат (NaHCO3), мочевина, тиомочевина, муравьиная кислота; Сегнетова соль (тартрат натрия-калия), 2,2-бицинхониновая кислота (2,2-БХК); коктейль ингибиторов протеаз (cOmplete™, Mini Protease Inhibitor Cocktail, «Sigma-Aldrich»); бычий сывороточный альбумин (БСА, «Sigma-Aldrich»); трипсин (Pierce MS Grade, «Thermo Scientific Promega», США); этилендиаминтетрауксусная кислота (ЭДТА, «BioChemica Applichem», Германия); 2-меркаптоэтанол, бромфеноловый синий, Coomassie Brilliant Blue R-250, додецилсульфат натрия (ДСН), дитиотреитол (ДТТ, «Acros Organics», Бельгия); глицерин («Acros Organics»); эфир полиоксиэтилена нонилфенола (NP-40); среда Вильямса; 10% фетальная бычья сыворотка (FBS; «Gibco», США); пенициллин-стрептомицин («Gibco»); инсулин («Gibco»); гидрокортизон («Gibco»); 1,8% диметилсульфоксид (ДМСО), ВМ-3 — Bifunctional cytochrome P450/NADPH-P450 reductase (cyp102A1) (предоставлен коллегами Института биоорганической химии НАН Беларуси). Культивирование и обработка клеток Клетки HepaRG были получены из коллекции Института биомедицинской химии имени В.Н. Ореховича. Замороженные криоконсервационные образцы разогревали и засевали в пластиковые культуральные флаконы объемом 75 см² (T75-флаконы), наполненные средой Вильямса Е, дополненной 10% эмбриональной бычьей сывороткой (FBS), инсулином (5 мг/л), глутамином (2 ммоль/л), гидрокортизона гемисукцинатом и антибиотиками (пенициллин/стрептомицин, 1%). Культивацию проводили в условиях насыщенной влажности и концентрации углекислого газа 5%, поддерживаемых инкубатором при постоянной температуре 37 °C. Дифференцировку клеток индуцировали добавлением в среду ДМСО в концентрации 2%. Смену среды осуществляли дважды в неделю. По завершении роста и дифференцировки клетки аккуратно отделяли от поверхности флаконов раствором трипсина (0.25%) с ЭДТА, промывали PBS, подсчитывали и объединяли в общую суспензию. Образцы центрифугировали, супернатант удаляли, а полученный осадок замораживали в жидком азоте и сохраняли при температуре –80°С до момента использования. Две пробирки, содержащие по 3,5 млн клеток, размораживали на льду и ресуспендировали в 500 мкл буфера солюбилизации: RIPA буфер, содержащий 0,1% (масса/объём) ДСН и 1% (масса/объём) NP-40 в 50 мМ Трис-HCl (pH 8.0), 150 мМ NaCl, 1 мМ ЭДТА, 10 мкл коктейля ингибиторов протеаз (протокол 2); 7М мочевину, 2М тиомочевину в 50 мМ Трис-HCl (pH 8,1) и 10 мкл коктейля ингибиторов протеаз (протокол 2). Клетки с буфером гомогенизировали в гомогенизаторе Поттера на льду. Затем образцы инкубировали в течение 30 мин при 4°С на орбитальном шейкере с вращением платформы 1000 об/мин. После инкубации каждый образец обрабатывали ультразвуком (на ледяной бане, время цикла 25х2 с с интервалом в 20-25 с для избежания денатурации), затем центрифугировали при 14000 g и 4°С в течение 30 мин. Супернатант каждого образца отбирали в отдельный эппендорф. Концентрацию общего белка в экстрактах HepaRG определяли методом с 2,2-БХК [11] на спектрофотометре 8453 UV-Visible («Agilent», США) с БСА в качестве стандарта. 1DE-гель концентрирование и трипсинолиз в геле После солюбилизации клеточных осадков HepaRG в RIPA буфере (протокол 1) и определения концентрации белка отбирали аликвоту экстракта, содержащую 67.5 мкг белка. К аликвоте добавляли 1 мкл суспензии ВМ-3 (7.5 мкг белка). Экстракт клеток проходил стадию 1DE-гель концентрирования в полиакриламидном геле согласно ранее опубликованному протоколу []. После завершения процедуры визуально определяли единственную белковую полосу, которую извлекали из геля с помощью ручного микротома, разрезали скальпелем на 10 кубиков размером 1 мм3 и проводили гидролиз трипсином непосредственно в геле, используя методику Shevchenko и соавт. [12]. Раствор, содержащий смесь триптических пептидов, извлечённых из геля, использовали для дальнейшего анализа методом жидкостной хроматографии с тандемной масс-спектрометрией (LC-MS/MS). Трипсинолиз в растворе К отобранной аликвоте экстракта, содержащей 67.5 мкг белка, после солюбилизации в буфере на основе мочевины и тиомочевины (протокол 2) добавляли 7.5 мкг BM-3. Инкубировали с 10 мМ ДТТ в течение 1 ч при 37°C. Алкилирование проводили с помощью 20 мМ IAA в течение 30 мин в темноте. Образцы разбавляли 50 мМ Трис-HCl так, чтобы конечная концентрация мочевины составляла 1.5 М. Добавляли трипсин в соотношении 1:50 (трипсин:белок) и проводили трипсинолиз в растворе в течение ночи при 37°C. Реакцию останавливали добавлением муравьиной кислоты (конечная концентрация 5%, масса/объём) и центрифугировали при 14000 g в течение 30 мин при комнатной температуре. Экстракты пептидов объединяли и использовали для LS-MS/MS анализа. LC-MS/MS протеомное профилирование Протеомный анализ осуществляли с использованием оборудования ЦКП “Протеом человека” при ИБМХ. Пептиды перед аналитическим разделением наносили на обогащающую колонку Accalaim μ-Precolumn (0.5 мм × 3 мм, размер частиц 5 мкм; «Thermo Scientific») при скорости потока 10 мкл/мин в течение 5 мин в изократическом режиме подвижной фазы Б (2% ACN, 0.1% муравьиной кислоты). Затем пептиды разделяли на колонке Acclaim Pepmap C18 (75 мкм × 150 мм, размер частиц 2 мкм; «Thermo Scientific») в градиентном режиме элюирования (система ВЭЖХ Ultimate 3000; «Thermo Scientific»). Градиент формировали подвижными фазами А (0.1% муравьиная кислота) и Б (80% ACN, 0.1% муравьиной кислоты) при скорости потока 0.3 мкл/минут. Колонку промывали 2% подвижной фазой Б в течение 5 мин, после чего линейно увеличивали концентрацию подвижной фазы Б до 35% в течение 65 мин, затем за 5 мин линейно увеличивали концентрацию фазы Б до 99%. После промывки колонки (5 мин, 99% Б) концентрацию Б снижали до начальных условий — 2% фазы Б за 5 мин. Общая длительность анализа – 90 мин. Масс-спектрометрический анализ проводили на гибридном орбитальном масс-спектрометре Orbitrap Fusion («Thermo Scientific») в режиме положительной ионизации в источнике NESI («Thermo Scientific»). Напряжение на эмиттере – 2.1 кВ. Панорамное сканирование проводили в диапазоне масс от 400 m/z до 1200 m/z, тандемное сканирование фрагментных ионов — от нижней границы 100 m/z до верхней границы, определяемой зарядным состоянием прекурсорного иона, но не более 2100 m/z. Для тандемного сканирования учитывали только ионы от z = 2+ до z = 6+ по зарядному состоянию. Максимальное число разрешенных для синхронной изоляции ионов в режиме МС2 не более 20. Максимальное время накопления прекурсорных ионов составило не более 50 мс, фрагментных ионов не более 110 мс. Поиск белков/пептидов в первичных raw-файлах проводили с помощью протеомной поисковой машины IdentiProt, основанной на алгоритме IdentiPy [13, 14], с параметрами поиска: база данных “Swiss_Prot” (SP, версия 1.4.2019, “.fasta” формат) для вида Homo sapiens с интегрированной для BM3 базой uniprotkb_Bacillus_megaterium_AND_revie_2025_02_27.fasta; расщепляющий фермент – трипсин; точность измерения теоретической и экспериментальной массы пептида – ±5 ppm; точность измерения теоретической и экспериментальной массы фрагментарных ионов – ±0.01 Да; значение зарядового состояния ионов пептида – “2+, 3+ and 4+”; количество возможных пропущенных участков расщепления трипсином – 1; фиксированная модификация – карбамидометилирование цистеина; вариабельная модификация – окисленный метионин. Поиск проводили по базе данных инвертированных и случайных последовательностей аминокислот (decoy), процент ложноположительных результатов (FDR) – ≤1%. Идентификацию белков проводили одновременно по всем RAW-файлам (всего 6), по три файла/технического повтора на каждый образец. В результате получали списки идентифицированных пептидов и белков в формате TXT, которые переводили в соответствующие Excel-таблицы. Статистическую значимость различий между сравниваемыми показателями определяли по критерию Стьюдента (t-критерий) для независимых выборок. Данные представлены в виде среднего значения и стандартного отклонения. Результаты оценивали как статистически значимые при пороговом уровне статистической значимости p-value<0.05. Для выявления корреляции использовали коэффициент ранговой корреляции Спирмена [15]. Значение коэффициента >0.7 принимали в качестве высокого уровня корреляции. Набор данных доступен в Mendeley Data [16]. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Выбор протокола пробоподготовки оказывает значительное влияние на результаты масс-спектрометрического анализа. Недавно нами была предложена методика, с применением буфера на основе ДСН для солюбилизации белков в комбинации с 1DE-гель концентрированием и трипсинолизом в геле (протокол 1) [10]. Данная методика позволяет удалить ДСН в ходе электрофореза в геле без разделения белков. Электрофорез останавливается до вхождения белков в разделяющий гель, в результате все белки концентрируются в виде одной полосы, которая подвергается трипсинолизу и дальнейшему MS-анализу. Методика позволяет уменьшить потери белков и сложность пробоподготовки и уже показала свою применимость в отношении нитей хориона [10] по сравнению с методикой, основанной на обработке тканей буфером на основе мочевины и трипсинолизом в растворе. Помимо этого, методика была эффективна в отношении культуры клеток кератиноцитов человека HaCaT [17]. В ходе настоящего исследования мы сравнили применимость предложенной методики пробоподготовки для культуры клеток HepaRG с протоколом, включающим экстракцию белков буфером на основе мочевины/тиомочевины с последующим трипсинолизом в растворе (протокол 2) (рис. 1). Оба протокола предполагают использование цельного экстракта клеток.
Сравнение протоколов осуществляли на основании количества идентифицированных белков и пептидов, степени покрытия аминокислотной последовательности (%), диапазона молекулярных масс белков, соотношения пептид-белок и сходимости технических повторов. Мы оценивали «глубину» трипсинолиза в геле, так как в случае протокола 1 все белки экстракта были сконцентрированы в одной полосе, что могло повлиять на доступность для трипсина и уменьшить количество пептидов для идентификации белков. Оценку проводили для обоих протоколов по количеству идентифицированных пептидов и покрытию аминокислотной последовательности внешнего белкового стандарта — рекомбинантного цитохрома P450 BM-3 (Bifunctional cytochrome P450/NADPH-P450 reductase, CYP102A1) из грамположительной палочки Bacillus Megaterium. Это гем-содержащий белок с молекулярной массой 118 кДа, участвующий в окислении жирных кислот и ксенобиотиков и обладающий монооксигеназной активностью []. Белок содержит одну полипептидную цепь, включающую 1049 аминокислотных остатка. Согласно базе UniProt цитохром P450 BM-3 не имеет общих пептидов с белками человека. В связи с этим все идентифицированные пептиды для BM-3 будут уникальными и его можно использовать для оценки эффективности трипсинолиза в присутствии белков клеточной культуры человека HepaRG. Количество общего белка, цитохрома P450 BM-3 и соотношение трипсин:белок для трипсинолиза в геле и трипсинолиза в растворе были одинаковыми. Комбинация 1DE-гель концентрирования и трипсинолиза в геле (протокол 1) позволила идентифицировать 82.7 ± 1.5 пептидов BM-3, тогда как с помощью трипсинолиза в растворе (протокол 2) было идентифицировано 76.0 ± 0.0 пептидов (табл. 1). Список идентифицированных пептидов BM3 представлен в дополнительных материалах (дополнительные материалы, табл. S1). Известно, что трипсин обеспечивает покрытие белковой последовательности в диапазоне 40-90 %, которое зависит от природы белка (например, мембранный или растворимый), сложности образца и применяемого метода пробоподготовки []. Идентифицированные пептиды обеспечили высокое покрытие аминокислотной последовательности BM-3, которое составило 87.0 ± 0.4 % (протокол 1) и 83.7 ± 0.5 % (протокол 2), что согласуется с данными литературы [18]. Наблюдаемые различия средних величин числа идентифицированных пептидов и покрытия аминокислотной последовательности статистически значимы (р<0.05).
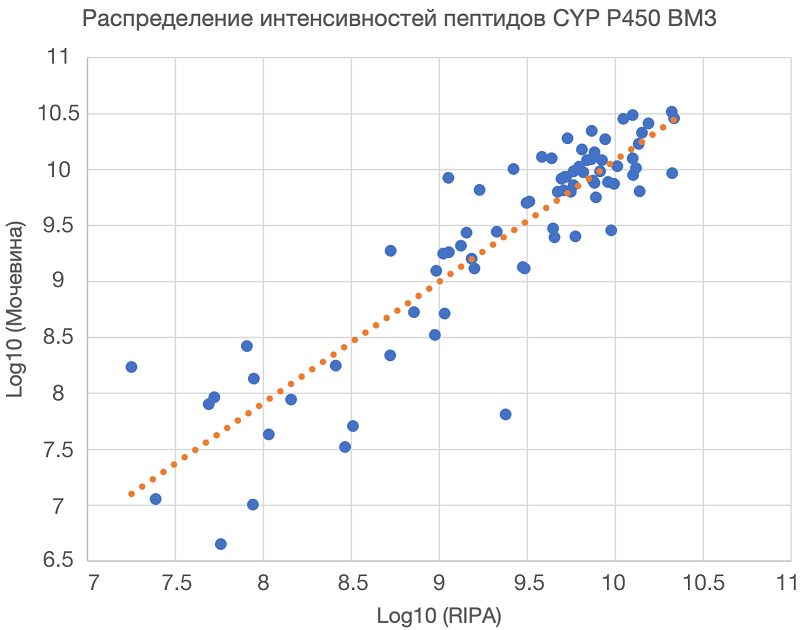
Важным критерием для оценки результатов протеомного анализа является воспроизводимость метода, о которой можно судить по сходимости результатов технических повторов. Для протокола 1 между техническими повторами было идентифицировано 75 общих пептидов, тогда как для протокола 2 — 73 общих пептида. Количество общих пептидов между двумя протоколами составило 73 пептида (83.9 % от общего числа идентифицированных пептидов), что свидетельствует об одинаковой доступности белка BM-3 для трипсина в геле (протокол 1) и в растворе (протокол 2) в присутствии сложной смеси белков HepaRG. Помимо количества идентифицированных пептидов и покрытия аминокислотной последовательности об эффективности трипсинолиза можно судить по интенсивности детектированных пептидов. Для оценки качества идентификаций и сравнения протоколов был рассчитан коэффициент корреляции Спирмена (r). Коэффициент корреляции может принимать значения от –1 до 1, причем при r = 1 имеет место строго прямая связь, а при r = –1 — строго обратная связь. На рисунке 2 представлено распределение интенсивностей общих зарегистрированных пептидов внешнего белкового стандарта для протокола 1 и протокола 2. По осям отложены значения десятичного логарифма интенсивностей пептидов. Как следует из рисунка, обе методики демонстрировали высокую корреляцию (r = 0.84) между значениями интенсивности зарегистрированных пептидов.
Таким образом, полученные результаты свидетельствуют о сопоставимой эффективности протоколов 1 и 2 в отношении внешнего белкового стандарта (цитохром P450 BM-3 из Bacillus megaterium) в присутствии экстрагированных белков культуры клеток HepaRG. При этом протокол 1 позволил идентифицировать большее количество пептидов и обеспечить более высокое покрытие аминокислотной последовательности цитохрома P450 BM-3 по сравнению с протоколом 2. На следующем этапе сравнение протоколов пробоподготовки проводили по количеству идентифицированных пептидов и белков клеточной культуры HepaRG и сходимости технических повторов. При использовании протокола 1 было идентифицировано 8487 ± 235 пептидов и 1242 ± 22 белков, при использовании протокола 2 количество идентифицированных пептидов и белков было несколько больше и составляло, соответственно, 9415 ± 276 пептидов и 1478 ± 34 белков (табл. 2). Список идентифицированных белков клеточной линии HepaRG представлен в дополнительных материалах (табл. S2 и S3). Всего было идентифицировано 3089 белков в экстракте клеток HepaRG, из них 2052 и 2589 белков для протоколов 1 и 2 соответственно. Различия в количестве идентификаций статистически значимы (р<0.05). Полученные нами результаты по количеству идентификаций согласуются с работой Tascher и соавт. [5], в которой в результате схожих методик пробоподготовки было идентифицировано 4394 белка в лизатах цельных клеток HepaRG. Разница в количестве идентифицированных в ней белков с нашими результатами может быть обусловлена методическими различиями в проведении экспериментов, например, условиями культивирования и масс-спектрометрии, соотношением трипсин:белок, а также используемой протеомной поисковой машиной для анализа данных.
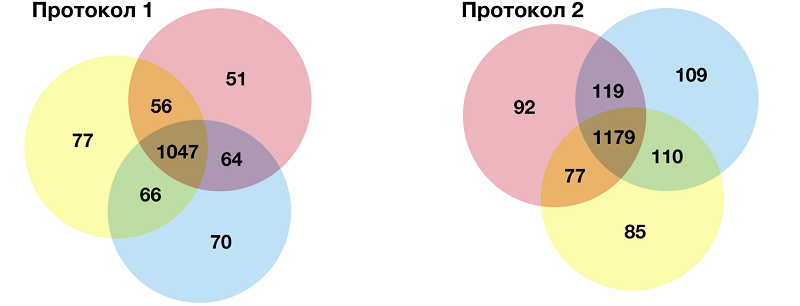
Несмотря на большее число идентифицированных белков, для протокола 1 соотношение пептид:белок оказалось выше (7.3 ± 3.0), чем при использовании протокола 1 (6.8 ± 3.0). Соотношение идентифицированных пептидов на белок отражает достоверность идентификаций, таким образом для протокола 1 качество идентификаций оказалось выше. Оба протокола пробоподготовки позволили идентифицировать белки в широком диапазоне молекулярных масс. Однако для протокола 1 диапазон молекулярных масс белков составил от 10 кДа до 629 кДа, в то время как протокол 2 позволил детектировать белки массой от 5 кДа до 629 кДа. При этом в обоих случаях большая часть белков (75%) находилась в диапазоне масс 10-100 кДа, что согласуется с данными литературы [19]. Сходимость результатов идентификаций технических повторов для протокола 1 оказалась несколько выше по сравнению с протоколом 2. На рисунке 3 представлены диаграммы Венна, отражающие распределение идентифицированных белков по техническим повторам. В случае протокола 1 сходимость результатов составила 71% или 1047 из 1468 белков, тогда как с помощью протокола 2 в трёх технических повторах было зарегистрировано 1179 общих белков из 1785, что составило 66% сходимости. Большая сходимость технических повторах в случае протокола 1 свидетельствует о надежности идентификаций.
На следующем этапе анализировали протеомы, полученные с помощью двух протоколов, на предмет наличия белков, специфичных для клеточной культуры HepaRG. В случае протокола 1 были идентифицированы такие референсные белки клеточной линии HepaRG, как succinate dehydrogenase flavoprotein subunit (SDHA), hypoxanthine-guanine phosphoribosyltransferase (HPRT1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 14-3-3 protein zeta/delta (YWHAZ), cytochrome c isoform 1 (CYC1), DNA topoisomerase 1 (TOP1), large ribosomal subunit protein uL13 (RPL13a) [20, 21]. Для протокола 2 детектировано меньшее количество референсных белков HepaRG: GAPDH, YWHAZ, CYC1, RPL13a. Помимо этого, с помощью протокола 1 были выявлены ферменты первого этапа метаболизма ксенобиотиков — цитохромы Р450 (CYP4F11, CYP27A1, CYP51A1), второго этапа метаболизма — h, UDP-glucuronosyltransferase 1A7 (UGT1A7), UDP-glucuronosyltransferase 2A2 (UGT2A2), UDP-glucuronosyltransferase 2A3 (UGT2A3) и thioredoxin (TRX), а также ферменты антиоксидантной защиты — g, glutathione reductase (GSHR) glutathione S-transferase A1 (GSTA1) и glutathione S-transferase Mu 1 (GSTM1). Как и в случае референсных белков, протокол 2 обеспечил идентификацию меньшего количества вышеперечисленных белков: CYP4F11, CYP51A1, HPT, TRX, GSH, GSTA1. Для того, чтобы более полно охарактеризовать эффективность протоколов, необходимы дополнительные эксперименты на основе дифференциальной экспрессии белков между интактной культурой клеток и культурой, обработанной ксенобиотиком. Настоящее исследование было осуществлено на контрольных образцах (не подвергшихся токсическому воздействию) клеток HepaRG. Для будущих работ, направленных на характеристику сравнительной эффективности протоколов, планируются работы по анализу дифференциальной экспрессии белков между интактной культурой клеток и культурой, после воздействия ксенобиотика. Так, например, наши предварительные показали, что 5-метилхолантрен практически не повлиял на протеомы клеток HepaRG. ЗАКЛЮЧЕНИЕ Наша работа посвящена оценке методики пробоподготовки, включающей солюбилизацию белков клеточной культуры HepaRG с помощью буфера на основе SDS (RIPA буфер) и очистки экстракта от детергента с помощью 1DE гель-концентрирования (протокол 1), в частности насколько полно можно охарактеризовать протеом данной культуры. В качестве методики сравнения был выбран протокол, включающий экстракцию с помощью 8М мочевины (протокол 2), в связи с тем, что эту методику используют для характеристики протеома клеточной линии HepaRG и она не предусматриваетпроведения дополнительных этапов перед процедурой трипсинолиза []. Протокол 1 и протокол 2 обеспечили схожее покрытие протеома клеточной культуры HepaRG, однако эффективность и воспроизводимость результатов идентификаций пептидов и белков зависела от способа пробоподготовки. Так, протокол 1 показал лучшее качество и достоверность идентификаций, а протокол 2 позволил идентифицировать большее количество белков. Кроме того, протокол 1 позволил идентифицировать больше референсных белков клеточной линии HepaRG. Для более полного покрытия протеома клеточной культуры HepaRG целесообразно использование обоих протоколов пробоподготовки. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов. БЛАГОДАРНОСТИ Масс-спектрометрические измерения были выполнены на оборудовании ЦКП “Протеом человека” Института биомедицинской химии (Россия). ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021–2030 годы) (No 122030100170-5). КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. К данной статье приложены дополнительные материалы, свободно доступные в электронной версии (http://dx.doi.org/10.18097/BMCRM00290) на сайте журнала. ЛИТЕРАТУРА
|