|
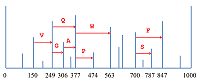
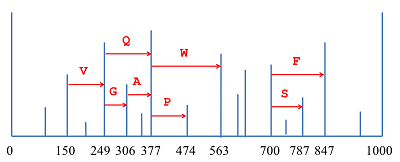
De novo секвенирование белков и пептидов: алгоритмы, приложения, перспективы Санкт-Петербургский национальный исследовательский Академический университет Российской Академии Наук; 194021, Санкт-Петербург, ул. Хлопина, д. 8, к. 3; тел.: (812) 448 69 80; факс: (812) 448 69 98; e-mail: vyatkina@spbau.ru Санкт-Петербургский государственный университет; 199034, Санкт-Петербург, Университетская наб., д. 7-9. Ключевые слова: апротеомика, масс-спектрометрия, de novo секвенирование, белки, пептиды, аминокислотная последовательность DOI: 10.18097/BMCRM00005 ВВЕДЕНИЕ Необходимость в исследовании структуры белков и пептидов возникает при решении самых разнообразных задач современной биологии и медицины. Ключевым его этапом является установление их аминокислотной последовательности, или секвенирование. Классическим методом, предложенным для этой цели, стала деградация по Эдману [1,2], суть которой заключается в циклически повторяющемся отщеплении меченого N-концевого аминокислотного остатка и его идентификации при помощи хроматографии. К основным факторам, ограничивающим ее применимость, относятся высокая стоимость реагентов и низкая скорость анализа. В частности, по этим причинам в настоящее время предпочтение нередко отдается более быстрому и менее дорогостоящему масс-спектрометрическому секвенированию. Установление первичной структуры белков или пептидов с использованием масс-спектрометрического метода сопровождается либо идентификацией масс-спектров посредством поиска в базе данных, либо их интерпретацией de novo. В первом случае необходимо наличие базы данных, предположительно содержащей аминокислотные последовательности изучаемых белков или пептидов. Второй же способ позволяет анализировать ранее неизвестные белки и пептиды, а также предоставляет дополнительные возможности для исследования аминокислотных последовательностей, содержащих неизвестные или многочисленные модификации. Алгоритмы идентификации масс-спектров путем поиска в базе данных разрабатываются на протяжении более двух десятилетий. В девяностых годах прошлого века были предложены, в частности, первые версии алгоритмов, ставших основой для коммерческих программных систем SEQUEST [3] и Mascot [4], широко используемых для идентификации пептидов и по сей день. К популярным программам, решающим ту же задачу и находящимся в свободном доступе, относятся, относятся, например, MS-GF+ [5,6] и Andromeda [7]. В течение последнего десятилетия были также разработаны и реализованы в программных инструментах подходы к идентификации белков по данным масс-спектрометрии «сверху вниз» (top-down), анализирующие белковые молекулы целиком; наиболее широкое распространение получила коммерческая реализация ProSightPC, сопровождаемая бесплатной Интернет-версией ProSight PTM [8,9], а также свободно распространяемые MS-Align+ [10] и TopPIC [11]. Следует отметить, что процесс идентификации масс-спектров по базе данных нередко включает в себя элементы de novo секвенирования: для ускорения поиска на предшествующем ему этапе может применяться фильтрация базы данных с использованием тегов пептидных последовательностей (peptide sequence tags) – коротких последовательностей аминокислот, представляющих собой результаты локальной интерпретации обрабатываемых масс-спектров. При этом в рассмотрении остаются лишь те аминокислотные последовательности из базы, которые согласуются с найденными тегами. Данный подход был впервые предложен в 1994 г. М. Манном и М. Вилмом [12], а впоследствии были опубликованы десятки статей, в которых обсуждались эффективные способы генерации тегов и их использования в качестве фильтров (см., например, [13-25] и [8]). Параллельно с методами идентификации по базе данных разрабатывались и алгоритмы de novo секвенирования. Первым алгоритмом, решающим данную задачу, стал Lutefisk97 [26], опубликованный в 1997 г. В основе этого метода, как и многих последующих, лежит концепция спектрального графа (spectrum graph), предложенная в 1990 г. Бартельсом [27]: такой граф сопоставляется масс-спектру, причем его вершины порождаются пиками, а ребра соединяют пары вершин, «отличающихся» друг от друга на массу остатка какой-либо аминокислоты (рис. 1). Каждое ребро спектрального графа помечено соответствующей аминокислотой (с точностью до замены I/L), а любой путь в этом графе определяет аминокислотную последовательность, образованную метками составляющих его ребер. Таким образом, полная или частичная интерпретация масс-спектра сводится к нахождению в его спектральном графе оптимального или нескольких лучших (с точки зрения используемой оценочной функции) путей.
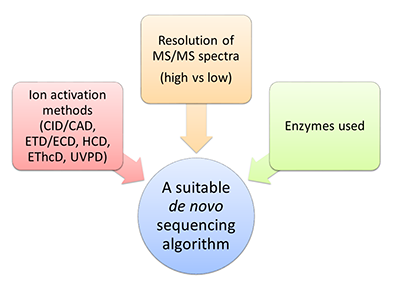
На сегодняшний день наиболее мощной и часто используемой коммерческой программой, несомненно, является PEAKS [28]; из бесплатных программных инструментов уже более десяти лет пользуется популярностью PepNovo [29]. К недавним разработкам относятся метод Twister, изначально предназначенный для de novo секвенирования пептидов по наборам тандемных масс-спектров «сверху вниз» (top-down) [30-32], а впоследствии адаптированный к случаю данных «снизу вверх» (bottom-up) высокого разрешения [33], и Novor [34], позволяющий обрабатывать масс-спектры триптических пептидов. Идентификация масс-спектров путем поиска в базе данных традиционно считается более надежным методом определения аминокислотной последовательности, нежели de novo секвенирование. Действительно, количество потенциально возможных интерпретаций масс-спектра, которые могут быть получены из базы данных, заведомо окажется существенно меньше числа всех возможных его интерпретаций noteПоясним данное утверждение на «игрушечном» примере. Пусть база данных состоит из трех последовательностей AN, TI и GEN массой 203, 232 и 318 Да. Соответственно. Допустим, что масса родительского иона для обрабатываемого масс-спектра составляет 203 Да. Тогда на основе базы данных для него может быть предложена лишь одна потенциально возможная интерпретация – AN, в то время как при de novo секвенировании a priori их будет пять: AN, NA, AGG, GAG и GGA. Какие именно из них будут проанализированы явно, и как будет выглядеть окончательный ответ, определяется деталями алгоритма и используемой функцией оценки надежности решения. , а, следовательно, значительно сократится и число неверных интерпретаций, что, в свою очередь, должно уменьшить риск ошибки при попытке выбрать единственный правильный вариант. Еще одна причина заключается в недостатке методов контроля качества результатов de novo секвенирования, сопоставимых с методами оценки уровня ложноположительных результатов (False Discovery Rate, FDR), используемых при поиске в базе данных [35]. Однако технологические достижения и алгоритмические разработки последних лет обеспечили повышение надежности методов de novo секвенирования, что открывает новые перспективы для их применения и позволяет эффективнее использовать их в тех случаях, когда идентификация по базе данных не представляется возможной. Физико-химические основы масс-спектрометрического метода анализа белков и пептидов детально изложены, например, в обзоре [36] и монографиях [37,38]. В данной же статье будет приведен краткий обзор существующих на сегодняшний день алгоритмов de novo секвенирования, а также задач, при решении которых они могут быть успешно использованы. В заключение будут обозначены дальнейшие пути их совершенствования и некоторые дополнительные области их применения. 1. АЛГОРИТМЫ За последние двадцать лет были предложены десятки алгоритмических подходов к решению задачи de novo секвенирования белков и пептидов. Подавляющее их большинство было разработано для установления первичной структуры пептидов по данным масс-спектрометрии «снизу вверх» (bottom-up). К тем из них, что оказались наиболее привлекательными для специалистов, могут быть отнесены Lutefisk [26,39], SHERENGA [40], PEAKS [28], PepNovo [29] и усовершенствованная его версия PepNovo+ [41-43], NovoHMM [44], Vonode [23], pNovo [45], UniNovo [46] и Novor [34]. PEAKS представляет собой коммерческую программную систему с дорогостоящей пользовательской лицензией; прочие перечисленные выше алгоритмы реализованы в программных продуктах свободного доступа. Суть метода, лежащего в основе PEAKS, заключается в генерации для каждого масс-спектра большого числа аминокислотных последовательностей – потенциальных его интерпретаций (в соответствии с исходной версией подхода их количество составляло 10000), и последующем выборе той из них, что наилучшим образом объясняет данный масс-спектр. NovoHMM и Novor опираются на методы машинного обучения и используют, соответственно, скрытые марковские модели (Hidden Markov Models, HMMs) и деревья решений (decision trees). В рамках остальных шести подходов нашла применение концепция спектрального графа [27]. Каждый из этих методов de novo секвенирования включает в себя функцию оценки надежности предлагаемой интерпретации масс-спектра. При определении такой функции или же при вычислении значений ее параметров явно или неявно принимаются во внимание свойства масс-спектрометрических данных, для обработки которых, в первую очередь, предназначен соответствующий алгоритм, что необходимо учитывать при выборе метода анализа данных конкретного эксперимента (рис. 2).
Так, ранние подходы используют свойства фрагментации молекулярных ионов при помощи диссоциации пептидных связей, индуцированной соударениями (ДИС; collision-induced dissociation, CID), или диссоциации, активированной соударениями (ДАС; collision activated dissociation, CAD), в то время как pNovo был разработан для анализа масс-спектров, полученных с использованием ДАС при повышенной энергии (ДАСПЭ; higher-energy C-trap dissociation, HCD) [47], предложенной в 2007 г. Однако более поздние версии алгоритмов интерпретации ДИС- и ДАС-МС/МС спектров, как правило, позволяют получать приемлемые результаты и для ДАСПЭ-МС/МС спектров. Последние версии PEAKS, наряду с ДИС- и ДАС-МС/МС спектрами, позволяют анализировать масс-спектры, полученные с помощью диссоциации при переносе электрона (ДПЭ; electron transfer dissociation, ETD) [48] или диссоциации при захвате электрона (ДЗЭ; electron capture dissociation, ECD) [49], а также комбинированного метода активации диссоциации EThcD (electron-transfer/higher-energy collision dissociation) [50] и ультрафиолетовой фотодиссоциации (УФФД; ultraviolet photodissociation, UVPD) [51], однако детали соответствующих алгоритмов не разглашаются. Прочие перечисленные выше алгоритмы не предназначены для обработки ДПЭ/ДЗЭ-, EThcD- или УФФД-МС/МС спектров. Отдельного упоминания в данном контексте заслуживает недавно предложенный подход UVNovo [52] к de novo секвенированию меченых хромофором триптических пептидов по наборам УФФД-МС/МС спектров; алгоритмическая его составляющая базируется на машинном обучении и включает в себя использование метода случайного леса и скрытых марковских моделей. При выборе подходящего метода de novo секвенирования существенное значение имеет и то, с низким или высоким разрешением были сняты MC/МС-спектры. Из обсуждаемых девяти алгоритмов лишь Vonode был изначально предназначен для обработки MC/МС-спектров высокого разрешения. Кроме того, к этому случаю адаптирована версия PepNovo+ алгоритма PepNovo, а также достаточно поздние версии программной системы PEAKS. В то же время, качество результатов применения к таким данным алгоритмов, ориентированных на обработку масс-спектров низкого разрешения, порой оказывается абсолютно неприемлемым. Так, в [33] приведены крайне бедные результаты обработки наборов масс-спектров, снятых с высоким разрешением на приборах Q-Exactive plus MS и Q-Exactive Orbitrap HF (Thermo Fisher Scientific, Бремен, Германия) при помощи метода Novor, способного весьма эффективно обрабатывать МС/МС-спектры низкого разрешения в режиме реального времени. И в противоположной ситуации, очевидно, не будет оснований рассчитывать на хорошие результаты. Наконец, следует иметь в виду, что практически все методы de novo секвенирования пептидов рассчитаны, прежде всего, на случай триптических пептидов. Из упомянутых выше программ лишь PEAKS предоставляет возможность явного указания других ферментов, использованных для гидролиза исследуемых белков, при выборе параметров эксперимента noteЗаметим, что скриншоты, иллюстрирующие возможности сервиса по de novo секвенированию белков, предоставляемого компанией Rapid Novor – разработчиком алгоритма Novor, доступные на сайте https://www.rapidnovor.com/demo/coverageview, содержат результаты для пептидов, полученных гидролизом шестью различными ферментами (трипсином, пепсином, химотрипсином, GluC, AspN и протеиназой К). Очевидно, закрытая версия Novor обладает соответствующей функциональностью, однако последняя его версия (1.05), находящаяся в свободном доступе, по-прежнему позволяет указать в качестве использованного фермента лишь трипсин. . Правда, многие алгоритмы предоставляют опции работы с «полутриптическими» или «нетриптическими» пептидами – однако при их выборе результаты интерпретации нередко теряют в аккуратности и надежности. Алгоритмы de novo секвенирования, реализованные в программах pNovo+ [53], ADEPTS [54] и UniNovo [46], а также методы, изложенные в работах [55-60], используют информацию из масс-спектров, снятых с одного и того же пептида с использованием различных методов инициирования фрагментации молекулярных ионов, что позволяет существенно улучшить покрытие последовательности, а, следовательно, и качество результатов. (Следует отметить, что UniNovo позволяет обрабатывать и наборы масс-спектров, полученные с использованием лишь одного из методов ДИС, ДАСПЭ или ДПЭ.) Также был разработан ряд подходов к de novo секвенированию белков по данным масс-спектрометрии «снизу вверх» для перекрывающихся пептидов, полученных гидролизом с применением нескольких ферментов различной специфичности или одного фермента без выраженной специфичности. Их суть заключается либо в выравнивании и объединении масс-спектров, предположительно относящихся к перекрывающимся пептидам, с последующим секвенированием полученного «суперспектра», либо в объединении результатов de novo секвенирования отдельных масс-спектров. Методы первого типа на этапе выравнивания масс-спектров могут не использовать [61,62] или использовать [63,64] гомологичные белковые последовательности. Алгоритм Champs, изложенный в [65], применяет PEAKS для интерпретации исходных масс-спектров и нахождения в базе данных последовательности белка, гомологичного исследуемому, выравнивает по отношению к этой последовательности полученные de novo теги, а затем предсказывает аминокислотную последовательность целевого белка путем ее уточнения. Наконец, повышение надежности результатов de novo секвенирования может быть обеспечено за счет сопоставления и комбинирования результатов, сгенерированных различными алгоритмами [66]. В завершение обсуждения подходов к de novo секвенированию пептидов следует упомянуть недавно предложенный разработчиками pNovo и pNovo+ алгоритм Open-pNovo [67], позволяющий учитывать потенциальное присутствие в последовательности любых из посттрансляционных модификаций, включенных в базу данных UniProt [68], а также выполненный в статье [69] анализ эффективности ряда методов в применении к смешанным (mixed) МС/МС-спектрам. Первый метод определения аминокислотной последовательности белка по данным масс-спектрометрии «сверху вниз» (top-down) был изложен в работе [70]; входными данными для него служат пары ДАС- и ДЗЭ-МС/МС спектров. В 2014 г. был предложен комбинированный метод TBNovo для de novo секвенирования белков по наборам масс-спектров «сверху вниз» и «снизу вверх» [71], в соответствии с которым вначале выполнялось de novo секвенирование пептидов по данным «снизу вверх» с применением программы PEAKS, а затем полученные пептидные последовательности объединялись в белковую с использованием «каркаса» (scaffold), сформированного на основе данных «сверху вниз». Первым же алгоритмом de novo секвенирования белков лишь по масс-спектрам «сверху вниз», реализованным в виде свободно распространяемого программного инструмента, стал Twister [30-32]. Основная его идея заключается в том, чтобы дать отдельным масс-спектрам частичную, но максимально надежную интерпретацию, а затем скомбинировать полученные таким образом теги пептидных последовательностей. Впоследствии этот подход был адаптирован к случаю данных масс-спектрометрии «снизу вверх» высокого разрешения [33]. Для того чтобы обеспечить возможность его применения к масс-спектрам «снизу вверх», необходимо предварительно их обработать при помощи метода деконволюции (удаления изотопных пиков и перехода от отношения массы к заряду к нейтральным массам), предназначенного для данных «сверху вниз». Результаты анализа, приведенного в [33], свидетельствуют о том, что предпочтительным является использование алгоритма MS-Deconv [72]. После этого в большей части MC/МС-спектров «снизу вверх» пиков останется совсем немного (не более 10-20) или не останется вовсе. Как следствие, индивидуальная интерпретация большинства из них окажется невозможной. Однако оставшиеся пики будут хорошо подтверждены и позволят сгенерировать аккуратные теги пептидных последовательностей, за счет комбинирования которых далее могут быть получены длинные фрагменты последовательностей анализируемых пептидов, а порой и полные их последовательности. Результаты предварительных экспериментов подтверждают возможность использования метода Twister и для обработки данных масс-спектрометрии «с середины вниз» (middle-down) высокого разрешения: в данном случае, речь идет о de novo секвенировании длинных пептидов, полученных путем ферментативного или химического гидролиза. Twister позволяет обрабатывать ДИС/ДАС-, ДПЭ/ДЗЭ- и ДАСПЭ-МС/МС спектры и может быть также применен к EThcD-МС/MC спектрам. При обработке масс-спектров «снизу вверх» и «с середины вниз» Twister не использует никаких предположений относительно того, какие ферменты или химические соединения использовались для гидролиза, что делает его более универсальным по сравнению с другими методами. 2. ПРИЛОЖЕНИЯ Алгоритмы de novo секвенирования востребованы, прежде всего, при изучении белков, которые не могут быть проанализированы на уровне генома или транскриптома, но они также находят применение и при исследовании сложных белковых смесей (рис. 3).
Наиболее распространенным приложением методов de novo секвенирования белков и пептидов, несомненно, является их использование для установления первичной структуры антител. Необходимость в этом возникает, в частности, при разработке лекарственных препаратов на основе моноклональных антител, которые, согласно оценке экспертов, в последние годы занимают наибольшую долю фармацевтического рынка среди биопрепаратов (см., например, [73]). Поэтому неудивительно, что многие алгоритмы были либо целенаправленно разработаны для решения этой задачи [63,64,74], либо протестированы на наборах масс-спектрометрических данных для антител [30-32,68], а компания Bioinformatics Solutions – разработчик системы PEAKS – летом 2017 г. выпустила на рынок программный пакет PEAKS AB [74], предназначенный для de novo секвенирования антител и анализа их аминокислотных последовательностей. Все эти алгоритмы предназначены для анализа либо отдельных моноклональных антител, либо простых их смесей, называемых «коктейлями» (antibody coctails), однако недавно была предпринята попытка разработать алгоритм, позволяющий анализировать поликлональные антитела [75]. Еще одним примером белков, при изучении которых возникает потребность в использовании методов de novoсеквенирования, являются токсины. Понимание их структуры и функций также необходимо для разработки ряда лекарственных средств, включая противоядия. Полноценное исследование токсинов методами геномики или транскритпомики не представляется возможным, во-первых, в силу недостатка необходимой для этого информации (в частности, единственной змеей, для которой полностью секвенирован геном и транскриптом ядовитой железы, остается королевская кобра [76,77]), а во-вторых, ввиду их подверженности посттрансляционным модификациям, оказывающим существенное влияние на их биологическую активность. В работе [62] приведены результаты анализа с применением изложенного в ней метода «скорострельного секвенирования белка» (shotgun protein sequencing) состава яда техасского гремучника (Crotalus atrox), а в статье [78] предложен метод de novo секвенирования «с ограничениями», предназначенный для анализа токсинов улиток-конусов (результаты исследования приведены для видов Conus textile и Conus stercusmuscarum). Ограничения формулируются на основе априорных знаний о последовательности токсина: это может быть, например, количество входящих в ее состав аминокислотных остатков цистеина или какие-либо «мотивы» (motifs), которые должны в ней присутствовать; введение их в рассмотрение позволяет существенно уменьшить число последовательностей-кандидатов, из которых выбирается окончательный ответ. Не менее интересным объектом исследований методами de novo секвенирования являются пептиды из кожного секрета амфибий, обладающие антимикробными, противоопухолевыми, противогрибковыми и другими свойствами [79] – в частности, кожные пептиды-антибиотики ранидных лягушек, различные виды которых широко представлены в России и Европе [80,81]. Структурной их особенностью является наличие C-концевого цикла, который возникает за счет образования дисульфидной связи между двумя цистеиновыми остатками; он носит название Rana box [81]. Для определения аминокислотной последовательности внутри такого цикла его необходимо предварительно раскрыть. Полученный в результате пептид будет иметь линейную структуру, однако, очевидно, не будет походить на триптический, что существенно снизит эффективность применения для его анализа практически всех существующих алгоритмов de novo секвенирования (напомним, что из обсуждавшихся выше методов возможности для анализа нетриптических пептидов предоставляют лишь поздние версии PEAKS и недавно предложенный алгоритм Twister, а также потенциально Novor.) Как следствие, ранее для установления первичной структуры таких пептидов либо применялась деградация по Эдману [82], либо de novo секвенирование выполнялось вручную [80]. Кроме того, алгоритмы de novo секвенирования белков и пептидов с успехом применялись для решения самых разнообразных специализированных задач – например, при изучении абиогенных пептидов [83], микробных сообществ [84], нейропептидома морского ежа [85], особенностей адаптации личинок к зимовке [86], а также в контексте других исследований. 3. ПЕРСПЕКТИВЫ К основным направлениям дальнейшего совершенствования подходов к de novo секвенированию белков и пептидов с целью расширения их применимости на практике относится повышение аккуратности выдаваемых ими результатов, а также разработка универсальных методов оценки их надежности. Для достижения первой цели при улучшении существующих и разработке новых алгоритмов следует в максимально возможной степени использовать современные достижения масс-спектрометрии высокого разрешения и учитывать особенности получаемых с ее помощью данных. В частности, представляет интерес разработка специальных алгоритмов для интерпретации весьма информативных, но сложных с точки зрения обработки УФФД- и EThcD-МС/МС спектров. Кроме того, найдут применение алгоритмы de novo секвенирования пептидов, полученных гидролизом ферментами без выраженной специфичности к конкретным аминокислотным остаткам (например, секретируемой аспарагиновой протеазой 9 [87,88]), а также эндогенных пептидов, включая пептиды-антибиотики амфибий [80,81]. Для оценки надежности результатов идентификации белков или пептидов посредством поиска в базе данных, как правило, оценивается уровень ложноположительных результатов (FDR), определяемый как отношение числа ложноположительных идентификаций к общему их количеству. Наиболее распространенной является следующая схема: на основе «целевой» (target) базы данных путем реверсирования каждой из входящих в нее последовательностей генерируется «ложная» (decoy), а затем выполняется поиск в объединенной базе данных [35]. При этом предполагается, что число ложноположительных идентификаций в целевой базе данных совпадет с числом (заведомо ошибочных) идентификаций в ложной. Далее критерии, на основе которых предложенные идентификации разделяются на «принятые» и «отклоненные», могут быть выбраны таким образом, чтобы обеспечить желаемый уровень FDR (например, 1%). В англоязычной литературе данный подход носит название «target-decoy approach». Очевидно, этот метод не может быть непосредственно применен в случае de novo секвенирования. Однако в недавних работах [89, 66] была продемонстрирована возможность получения аккуратной оценки FDR для результатов de novo секвенирования с использованием результатов идентификации в базе данных. Также представляет интерес опубликованный в январе 2018 г. метод оценки уровня «ложных аминокислот» (false amino-acid rate, FAR) [90], определяемого как отношение числа неправильно предсказанных аминокислот к общему количеству аминокислот в последовательностях, сгенерированных алгоритмом de novo секвенирования. В то же время не вызывает сомнений, что исследования в данном направлении будут продолжаться. ЗАКЛЮЧЕНИЕ Методы de novo секвенирования представляют собой незаменимый инструмент анализа белков из организмов, геном которых неизвестен, а также тех белков, которые напрямую не закодированы в геноме – например, антител. Достижения современной масс-спектрометрии высокого разрешения открывают новые возможности для их совершенствования, а, вместе с тем, расширяется и круг задач, для решения которых они могут быть с успехом использованы. Неудивительно, что в ведущих научных журналах регулярно появляются новые публикации, посвященные разработке и применению алгоритмов de novo секвенирования, все более востребованных как при изучении отдельных белков и пептидов, так и в комплексных протеогеномных исследованиях. БЛАГОДАРНОСТИ
Работа выполнена при поддержке Российского фонда фундаментальных исследований (грант №16-54-21006).
ЛИТЕРАТУРА
|