К 40-летию Института физиологически активных веществ РАН
|
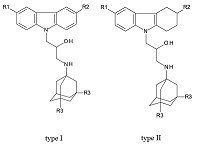
QSAR моделирование блокады NMDA рецептора полифармакофорными соединениями на основе производных карбазола и 1-аминоадамантана Институт физиологически активных веществ Российской академии наук 142432 Черноголовка Московской обл., Северный проезд, 1; *e-mail: beng@ipac.ac.ru Ключевые слова: QSAR; NMDA рецептор; полифармакофорные соединения DOI: 10.18097/BMCRM00064 ВВЕДЕНИЕ Одним из вызовов современности, который требует быстрого и адекватного ответа, является необходимость разработки медицинских препаратов для лечения нейродегенеративных заболеваний, в частности, болезни Альцгеймера (БА) [1]. В настоящее время выявлен ряд биологических мишеней, связанных с этой болезнью [2]. Одной из них является NMDA рецептор [3], играющий важную роль в функционировании центральной нервной системы. Одним из перспективных направлений при разработке лекарств для БА является использование соединений с несколькими фармакофорными группами, действующими на несколько биомишеней одновременно [4]. Целью настоящей работы является конструирование регрессионных моделей блокады NMDA рецептора (сайт связывания [H3]MK-801) полифармакофорными соединениями на основе производных карбазола и 1-аминоадамантана. МАТЕРИАЛЫ И МЕТОДЫ Выборка, содержащая информацию о структуре и биологической активности соединений, была извлечена из базы данных [5]. Объектами исследования служили 14 соединений, представляющих собой конъюгаты замещенных карбазолов (тип I) и тетрагидрокарбазолов (тип II) с 1-аминоадамантанами (рис. 1, табл. 1).
В качестве первоначальной количественной оценки биологической активности использовали величину IC50 (мкМ) (концентрация при которой наблюдается 50% торможение исследуемым соединением связывания [H3]MK-801 с NMDA рецептором). Для QSAR моделирования эти величины были конвертированы в -lg(IC50). При этом минимальное, максимальное и среднее значение активности в анализируемой выборке составили -2.217, -1.190 и -1.929 соответственно. Для количественного описания структуры соединений рассчитали несколько сот физико-химических, топологических, электронных дескрипторов с использованием компьютерных программ HYBOT [6] и DRAGON [7]. В качестве следующего шага была применена процедура сокращения дескрипторного пространства на основе анализа парных коэффициентов корреляции (rij) при критическом значении r = 0.8. В результате число дескрипторов было уменьшено до 34. В состав финальных QSAR моделей вошло всего 7 дескрипторов: BAC (центрированный индекс Балабана), CIC4 (комплементарное информационное содержание 4-го порядка), MATS7v (автокорреляция Морана с лагом 7, взвешенная атомными ван-дер-ваальсовыми объемами), MATS8v (автокорреляция Морана с лагом 8, взвешенная атомными ван-дер-ваальсовыми объемами), EEig10x (10 собственное значение матрицы связности, взвешенное по степеням вершин), BEHe4 (4 высшее собственное значение матрицы Бурдена, взвешенное атомными электроотрицательностями Сандерсона), Psy-80 (индекс Ghose-Viswanadhan-Wendoloski антипсихотического типа при 80%). Таблица 1 содержит данные, которые были использованы для проведения QSAR моделирования.
QSAR моделирование проводили с использованием трех методов: линейной регрессии (MLR) [8], случайного леса (RF) [9] и гауссовского процесса (GP) [10]. Для оценки предсказательной способности регрессионных моделей применяли перекрестный контроль с выбором по 5. В качестве статистических характеристик моделей использовали: n – число соединений; m – число дескрипторов; R2 – квадрат коэффициента линейной корреляции; rmse – среднеквадратичное отклонение; R2cv – квадрат коэффициента линейной корреляции в условиях перекрестного контроля; rmsecv - среднеквадратичное отклонение в условиях перекрестного контроля; R2p – метрика для оценки случайной корреляции [11]. Область применимости (ОП) моделей оценивали на основе анализа интервалов величин дескрипторов. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ В таблице 2 приведены результаты проведенного QSAR моделирования блокады NMDA рецептора. Из представленных данных ясно, что регрессионные модели MLR и RF не могут быть использованы для предсказания активности новых соединений из-за своих низких статистических показателей. Но модель, созданная на основе Гауссовского процесса, выглядит вполне адекватной. В состав модели входит 2 дескриптора, что является вполне приемлемым, так как (n/m) > 5. Величина R2 > 0.6, R2cv > 0.5 и R2p > 0.5, что соответствует требованиям, предъявляемым к QSAR моделям [11,12]. ОП модели (интервальная оценка): MATS7v – -0.407 ÷ 0.165; EEig10x – 2.537 ÷ 3.137. Что касается физико-химического содержания дескрипторов GP модели, то достаточно трудно дать им однозначное толкование. Можно лишь отметить, что они являются топологическими дескрипторами и легко могут быть рассчитаны для различных молекул.
Таким образом, анализ полученных результатов свидетельствует о том, что способность исследуемых соединений влиять на связывание [H3]MK-801 с NMDA рецептором может быть удовлетворительно описана с помощью регрессионной модели на основе Гауссовского процесса с небольшим числом дескрипторов. Эта статистическая модель может быть использована для количественной оценки биоактивности новых полифармакофорных соединений на основе производных карбазолов и тетрагидрокарбазолов с 1-аминоадамантанами. БЛАГОДАРНОСТИ Работа выполнена в рамках государственного задания на 2018 год (тема № 0090-2017-0020). ЛИТЕРАТУРА
|