|
Исследование канальных блокаторов NMDA рецептора в ряду конъюгатов метиленового синего с использованием QSAR и молекулярного моделирования
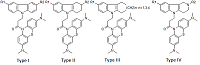
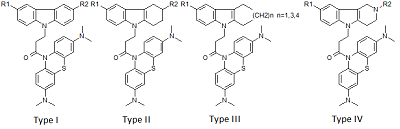
1Институт физиологически активных веществ РАН, Ключевые слова: NMDA рецептор; канальные блокаторы; QSAR; докинг DOI: 10.18097/BMCRM00091 ВВЕДЕНИЕ В настоящее время в мире различные формы деменции, в частности болезнь Альцгеймера (AD), диагностированы у 30 миллионов человек и опубликованы малоутешительные прогнозы о том, что их число может достичь 100 миллионов к 2050 году [1], по некоторым оценкам экономические потери в обозримом будущем могут превысить 1 триллион долларов [2]. Возможность лечения AD или улучшения качества жизни больных остаётся важной и пока не решённой проблемой, требующей создания новых лекарственных препаратов, в том числе с использованием вычислительных технологий [3]. В этой области, помимо общих проблем разработки новых лекарств (например, оптимизации связывания молекул с биомишенями и ADMET свойств), возникают дополнительные сложности, обусловленные мультифакторной природой патогенеза AD. Перспективным путём считается поиск новых соединений, действующих одновременно на несколько биомишеней [4-6], в частности, на ионотропные глутаматные NMDA рецепторы (блокирующих их действие). Опубликован ряд работ, посвященных изучению связывания лигандов с NMDA рецептором с использованием методов QSAR и молекулярного моделирования [7-9]. В тоже время известно о нейропротекторной активности таких соединениий как димебон и его аналоги [10], а также метиленового синего [11]. Целью настоящего исследования было создание моделей блокады NMDA, использующих методы QSAR и молекулярный докинг с NMDA рецептором (сайт связывания MK-801) и описывающих связывание гибридных бинарных структур, объединяющих свойства данных соединений МАТЕРИАЛЫ И МЕТОДЫ Сведения о структуре и биологической активности соединений были собраны ранее и депонированы в специализированной базе данных [12]. В работе использованы 29 химических соединений – конъюгатов восстановленной формы метиленового синего (МB) и четырех типов соединений, включая производные карбазола (I), тетрагидрокарбазола (II), замещенных индолов (III) и γ-карболина (IV), объединенных 1-оксопропиленовым спейсером (рис. 1).
В качестве меры биологической активности соединений служила величина IC50 (мкМ), которую определяли с использованием радиолигандного метода на основе изучения влияния соединений на связывание [H3]MK-801 с NMDA рецепторами, полученными из мембран гиппокампа крыс [13]. Для описания исследуемых соединений набором дескрипторов была использована программа HYBOT, в основе которой лежит расчет количественных характеристик водородной связи [14] и некоторых других свойств молекул. В состав конечных регрессионных моделей вошло суммарно 4 дескриптора, включая: α (молекулярная поляризуемость), Σ(Ca) (сумма свободноэнергетических Н-акцепторных дескрипторов), Σ(Ca)/α (композитный дескриптор), Eamax (максимальный энтальпийный Н-акцепторный дескриптор). Описание соединений и величины дескрипторов, использованных для расчетов, приведены в таблице 1.
При создании QSAR моделей, описывающих биологическую активность, использовали четыре метода: линейную регрессию (MLR), случайный лес (RF), опорные вектора (SVM) и гауссовский процесс (GP). Оценку регрессионных коэффициентов и определение степени коллинеарности переменных проводили компьютерной программой SVD [15]. Для анализа деревьев решений в методе RF использовали оригинальную программу автора метода Breiman [16] с исходными параметрами и числом деревьев, равным 500. В качестве машины опорных векторов применяли алгоритм flssvm [17], особенностью которого является использование метода наименьших квадратов. Конструирование GP моделей выполняли с применением алгоритма, описанного в работе [18]. Оценку предсказательной способности QSAR моделей проводили на основе внутреннего (кросс-валидация) и внешнего тестирования. Кросс-валидацию осуществляли путем случайного разбиения анализируемой выборки на пять равных частей, используя четыре части для конструирования модели и одну часть для ее тестирования (всего 5 вариантов). Тестовая выборка была сформирована путём упорядочивания исследуемых соединений по активности и выбору каждого пятого соединения. Таким образом, была создана обучающая выборка (24 соединения) и тестовая выборка (5 соединений). Для описания статистических свойств регрессионных моделей использовали: r2 – квадрат коэффициента линейной корреляции (обучающая выборка); rmse – среднеквадратичное отклонение (обучающая выборка); r2cv – квадрат коэффициента линейной корреляции в условиях кросс-валидации; rmsecv - среднеквадратичное отклонение в условиях кросс-валидации; r2test – квадрат коэффициента линейной корреляции (тестовая выборка); rmsetest - среднеквадратичное отклонение (тестовая выборка); r2p – метрика для оценки случайной корреляции, которую вычисляли по формуле [19]:
Молекулярный докинг соединений был проведен в структуру NMDA рецептора (NMDAR) XenopusLaevis (код PDB 5uow). Данный рецептор состоит из 4 субъединиц: 2 GluN1 (цепи А, С), GluN2A(цепь В) и GluN2B (цепь D). Хотя измерение активности исследованных соединений был проведено на NMDAR крысы, использование для докинга NMDAR лягушки было обусловлено следующими причинами: 1) данных о структуре NMDAR R. Norvegicus отсутсвуют, в то время как пространственная структура NMDAR XenopusLaevis в комплексе с МК-801 известна; 2) выравнивание аминокислотных последовательностей участков, образующих пору ионного канала, показало высокую степень их идентичности, что позволяет ожидать получение адекватного результата при использовании NMDAR X. Laevis вместо NMDAR R. Norvegicus. При подготовке структуры NMDAR X. Laevis к вычислениям были достроены недостающие в файле 5uow участки аминокислотной последовательности с использованием пакета SYBYL 8.1. Структура полученного комплекса была оптимизирована с использованием процедуры минимизации энергии средствами SYBYL. Докинг осуществляли программой AutoDock Vina [20], подготовку белка и лигандов -программой AutoDock Tools [21]. Место связывания в белке было определено как бокс размером 20х20х40Å, вмещающий всё пространство ионного канала, включая место связывания молекулы МК-801 в комплексе. Для каждого лиганда были получены девять вероятных поз докинга. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ В таблице 2 представлены параметры построенных QSAR моделей, характеризующих торможение (блокаду) NMDA рецептора исследуемыми соединениями. Линейная модель (MLR) показывает худшие результаты по сравнению с нелинейными моделями (RF, SVM, GP). При этом нелинейные QSAR модели удовлетворяют минимальным требованиям, которые к ним предъявляются [22], в частности, r2 > 0.6, r2cv > 0.5, r2test > 0.5, r2p > 0.5.
Об удовлетворительной предсказательной способности анализируемых моделей свидетельствуют также результаты предсказания для тестовой выборки (табл. 3). В 4 из 5 случаях (за исключением соединения 24) разность между экспериментальными и рассчитанными величинами активности не превышает величины 2-х стандартных отклонений по отношению к rmsecv. В случае соединения 24 наблюдаются систематические отклонения величин Аcalc–Аpred. Одна из возможных причин этого связана с тем, что в анализируемой выборке соединений присутствуют как соединения в нейтральной форме, так и в виде солей, что не принимается во внимание при расчете дескрипторов из-за особенностей работы программы HYBOT.
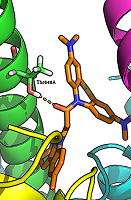
Следует отметить, что современные QSAR модели должны иметь не только хорошие статистические характеристики, но и обладать (по возможности) способностью к физико-химической интерпретации [23], т.е. давать возможность установить понятные причинно-следственные связи при описании исследуемого явления. В построенных моделях в качестве независимых переменных в основном работают дескрипторы водородной связи. При этом наиболее значимой величиной является Н-акцепторная способность исследуемых соединений. Данное наблюдение хорошо согласуется с имеющимися представлениями о том, что Н-связь играет важную роль при блокаде ионных каналов NMDA рецептора [24]. Полученные статистические модели содержат минимальное число дескрипторов, в то время как представленные в литературе QSAR модели, описывающие блокаду NMDA рецептора на основе количественных данных по вытеснению [3H]MK-801 из его сайта связывания, включают в свой состав 10 и более дескрипторов [8, 25] с неясной физико-химической интерпретацией. Анализ полученных результатов свидетельствует о том, что между активностью исследуемых соединений и их структурой обнаруживается простая линейная тенденция, которая заключается в том, что с увеличением протоноакцепторной способности соединений также растет их способность к связыванию с NMDA рецептором. Это становится очевидным при сопоставлении величин активности соединений I, II, и III типа с соединениями IV типа: появление в структуре дополнительного атома азота приводит к увеличению Н-акцепторной способности и, как следствие, ведет к увеличению активности. Для исследования механизма связывания этих соединений с NMDAR было проведено моделирование комплексов (докинг в ионный канал рецептора). Результаты моделирования показали, что исследуемые соединения располагаются в ионном канале NMDAR именно в месте связывания известного антагониста MK-801 (дизоцилпин). Исследуемые лиганды взаимодействуют с ионным каналом в основном за счет гидрофобных взаимодействий. В анализируемых комплексах также ожидаемо наблюдается благоприятное взаиморасположение лигандов и белка для образования водородных связей, обеспечивающих специфическое узнавание лиганда. Основной водородной связью, наблюдаемой у всех лигандов, является связь между карбонильным кислородом лиганда и ОН-группой Thr646 А-цепи GluN1 (рис. 2). Это отличает их от ранее известных антагонистов, связывающихся с Asn614 субъединицы GluN1, Asn602/Asn603 субъединицы GluN2A и Asn608/Asn609 субъединицы GluN2B [26].
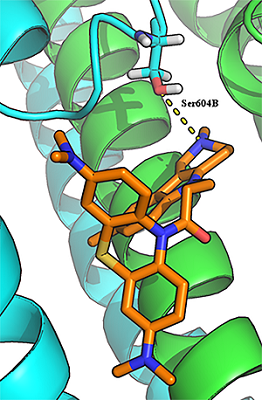
Для лигандов, содержащих дополнительный (пятый) атом азота (соединения 23-29), наблюдали дополнительный паттерн связывания. Дополнительный атом азота мог образовывать водородную связь с гидроксилом Ser604B-GluN2A (рис. 3). Возможность образование данной Н-связи может объяснить более низкое (лучшее) значение IC50 этой группы соединений по сравнению с остальными лигандами.
Таким образом, результаты молекулярного докинга хорошо согласуются с результатами, полученными при QSAR моделировании. Установлено, что водородные связи играют важную роль в связывании конъюгатов метиленового синего с NMDAR, а появление в структуре дополнительного атома азота приводит к увеличению активности таких соединений. ФИНАНСИРОВАНИЕ РАБОТЫ Работа выполнена в рамках государственного задания ИФАВ РАН на 2019 год (тема № 0090-2019-0004). ЛИТЕРАТУРА
|