|
Предсказание значения изоэлектрической точки пептидов и белков с широким спектром химических модификаций
Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, Ключевые слова: пептид; изоэлектрическая точка; посттрансляционные модификации; химические модификации, предсказание свойств DOI: 10.18097/BMCRM00161 ВВЕДЕНИЕ
Изоэлектрическая точка (pI) – важная физико-химическая характеристика пептидов и белков, которая широко используется в практике современного эксперимента [1-2]. Например, в области протеомных исследований наиболее часто используются такие методы, как двумерный (2D) гель-электрофорез для белков и фракционирование пептидов [3-6] с использованием изоэлектрического фокусирования (IEF) для последующего анализа масс-спектрометрическими методами. Кроме того, значение pI может быть использовано как дополнительный маркер при контроле правильности идентификации пептидов, реже – белков (так как белки в значительной степени подвергаются посттрансляционной модификации (PTM), то определение, какая конкретно протеоформа исследуется, – само по себе часто непростая задача [7]). Для предсказания значения pI обычно используют два методических подхода. Самый распространённый – использование уравнения Хендерсона-Хассельбаха [8]. Как правило, в случае пептидов и белков используют шкалу значений констант диссоциации (pKa) для отдельных химических групп, рассчитанную заранее. Самой популярной является шкала из работы [9], на базе которой создан широко используемый калькулятор [10] pI белков на Swiss Bioinformatics Resource Portal. Несмотря на то, что авторы [9] прямо пишут о предсказании значения в заданной области pH (от 4 до 7), за давностью лет этот факт уже забылся. Как правило, не учитывают и зависимость величины pKa от температуры и ионной силы раствора (справедливости ради, Chemaxon Marvin Suite [11] учитывает температурную зависимость для предсказания pKa). В тоже время, для белков, в которых потенциально заряженных групп достаточно много, ошибка, вносимая выбранной шкалой, обычно, невелика и лежит в пределах до 0.5 единиц pH. Для широкого спектра задач такая точность может оказаться достаточной. Однако в случае пептидов с ограниченным числом диссоциирующих групп эта ошибка может быть существенно выше. Существует несколько вариантов формирования шкалы значений pKa: квантово-химические расчёты (точность и время существенно зависят от сложности системы) и на основе эмпирических данных, полученных различными методами. Первый – проведение титрования модельных пептидов [12]. Это самый надёжный метод. Однако он долог, дорог и при использовании небольших выборок не даёт гарантии, что промоделированы все возможные комбинации. К тому же, число вариантов модельных структур растёт экспоненциально по мере увеличения длины пептида. Второй – решение обратной задачи: если для набора пептидов (белков) известны значения pI, то значения pKa подбираются таким образом, чтобы значение средней или среднеквадратичной ошибки было минимальным [13] (математический метод оптимизации параметров может быть и другим, вплоть до простого перебора значений). Для последнего способа нужно иметь набор большого числа пептидов с известными значениями pI. Такие наборы данных доступны как «побочный продукт» «shotgun»-протеомики с использованием фракционирования при помощи изоэлектрического фокусирования и широко применяются [2, 14, 15]. В случае их использования необходимо учитывать следующее:
Из положительных сторон данного метода следует отметить, в первую очередь, большой размер выборок, а также наличие в выборках модифицированных пептидов. Это позволяет сформировать шкалу, учитывающую посттрансляционные модификации, что встречается в очень ограниченном числе случаев (например, фосфорилирование у [16] или химическая модификации с использованием TMT (Tandem mass tag) и iTRAQ (Isobaric tag for relative and absolute quantitation) [14]). Кроме уравнения Хендерсона-Хассельбаха существует и другой подход для предсказания значения pI – применение методов машинного обучения с использованием генетических алгоритмов [17], искусственных нейронных сетей [13] и метода опорных векторов (SVM) [15, 18]. Однако, несмотря на кажущуюся привлекательность и несомненные достоинства этих методов, и у них также есть ряд ограничений и недостатков (переобучение выборки, невозможность предсказывать pI для полипептидов, существенно отличающихся по размеру от тех, что использовали в обучающей выборке, невозможность адаптации модели при введении новых вариантов PTM без полного пересчёта и др.). Что-то можно решить, используя отдельные модели для пептидов и белков, но для данных по белкам проблем ещё больше, чем для пептидов. Обычно значения pI для белков берут из данных, полученных при 2D электрофорезе. При этом часто неизвестно, о какой протеоформе идёт речь. Экспериментаторы не утруждают себя внесением маркёров pH, а привязку к шкале проводят либо по известным белкам, либо по предсказанным значениям, используя калькулятор pI с сайта Swiss Bioinformatics Resource Portal. Нередко публикуются данные, которые являются компиляцией из нескольких объединённых экспериментов, при этом в местах «склеек» могут быть ошибки. В любом случае, наборы данных существенно меньше по размеру, чем для пептидов. Данная работа посвящена формированию шкалы значений pKa, с использованием которой можно предсказывать величину pI как для пептидов, так и белков с широким спектром PTM и химических модификаций, как преднамеренных, так и спонтанных, наблюдаемых при проведении масс-спектрометрического анализа. МЕТОДИКА Формирование обучающей выборки пептидов Для формирования обучающей выборки были использованы данные экспериментов по масс-спектрометрической идентификации с примененим IEF, депонированные в БД ProteomeXchange (accession codes: PXD000065 [14], PXD005410 [19], PXD006291 [20], PXD010006 [21] и PXD017201 [22]). Всего было обработано 25 наборов данных (табл. 1), однако не все они были использованы для подбора обучающей выборки по причинам, указанным ниже. Так как для данной работы было важно получить максимально возможный спектр модификаций, что не было задачей авторов экспериментальных работ, то идентификацию пептидов провели заново с использованием программы Peaks Studio X Pro [23]. При этом, в качестве основных модификаций задавали химические модификации, специфичные для конкретного эксперимента (алкилирование цистеинов, TMT или iTRAQ метки), фосфорилирование, окисление метионина и дезаминирование. Остальные модификации находили, используя соответствующий модуль программы. Ограничение для идентификации прекурсора было установлено в 5 ppm, точность идентификации фрагментов – 0.01 Da. Были отобраны пептиды с уровнем FDR 5%. В ходе работы анализировали также варианты отбора с уровнями FDR 1% и 0.1%, но при резком сокращении числа вариантов существенного улучшения качества выборки после отбора не наблюдалось. Как правило, каждый идентифицированный пептид мог встречаться в нескольких пробах, ассоциированных с различными значениями pI. Это может быть связано как с ложной идентификаций, так и с тем фактом, что пептид встречается в нескольких соседних пробах с максимумом количества молекул в пробе c pH, наиболее близкой к pI пептида. Поэтому в результирующую выборку отбирали только те варианты, для которых максимум оценочной функции при идентификации пептида совпадал с максимумом площади под кривой распределения иона-прекурсора на хромотограме (данная величина может служить мерой количества пептида). Всего на первоначальном этапе было отобрано 613339 вариантов для 372215 неповторяющихся пептидов (табл. 1 и дополнительные материалы).
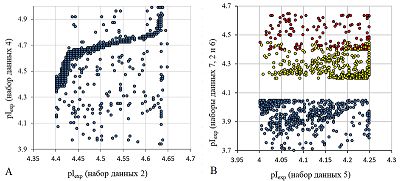
Для каждого пептида, идентифицированного более чем в одном наборе данных, разброс значений ΔpI колебался от 0 до 8 единиц pH. Частично это связано с тем, что реальные значения pH в каждом конкретном эксперименте могут отличаться от тех, что заявлены производителем стрипов для IEF, на которых происходит разделение пептидов по pH. В работе [14] один из наборов данных был получен с использованием pI маркеров. Таким образом, было возможно скорректировать значения pI, используя данные для пептидов, встречающихся и в данном наборе, и в том, для которого требуется коррекция. Исключение составил набор 15, его удалось выровнять по другим наборам данных (использовали набор 16, полученный теми же авторами). Наборы данных 13 и 17 (заявленный диапазон pH 2.5-3.7) из-за отсутствия значимых пересечений с другими наборами использовали без выравнивания. Следует отметить, что наличие одинаковых пептидов в наборах данных, полученных без облигатных химических модификаций, и с внесёнными метками (либо наборов с различными метками) скорее артефакт пробоподготовки. Ожидается, что все пептиды модифицированы, тем не менее в пробах всегда присутствует некоторая доля непрореагировавших пептидов, и если количество исходного пептида велико, то этой доли может хватить для его идентификации в неизменённом виде. Конечный отбор для формирования обучающей выборки проводили с использованием, следующих правил: пептид должен был быть идентифицирован в двух и более наборах данных, при этом, если хотя бы один из наборов имел диапазон значений pH от 3 до 10, расхождение по значениям pI должно было быть не более чем 0.15 единицы pH, для диапазонов pH 6-9 и 6-11 – не более 0.1, в случае если совпадение было только в наборах данных с диапазонами pH 3.7-4.9 и уже – не более 0.05. Из сравнения были исключены данные наборов 2, 5, 6 и 7 с шириной диапазона значений pH менее 0.5. Причиной стали наличие нелинейных зависимостей при попарном сравнении наборов (рис. 1А), а также тот факт, что они содержали слишком большое количество пептидов, для которых значение pI, определённое в других наборах, не совпало с заявленным диапазоном даже приблизительно. Более того, несмотря на то, что сами по себе эти 4 набора закрывают последовательно 4 диапазона значений pH c небольшим пересечением по границе, значительная часть пептидов находится более чем в одном наборе и часто более чем в двух (рис. 1B).
Для каждого конкретного пептида за значение pI принимали медианное значение по всем существующим вариантам. Сформированная обучающая выборка содержит 73863 пептида (см. дополнительные материалы); распределение значений pI и количество пептидов с наиболее представленными модификациями показаны на рисунке 2 (полностью статистика по всем модификациям приведена в дополнительных материалах).
Формирование шкалы значений pKa и тестирование В ходе работы были исследованы два варианта шкалы значений pKa. Первый, аналогичный шкале, опубликованной нами ранее [13], в которой значения pKa различаются для остатков, расположенных на N- и C-концах пептида и остатков со второго по предпоследний (обозначим её как шкала 3). Второй вариант предполагает учёт влияния соседних остатков. В идеале следует проварьировать все возможные варианты соседних остатков, однако для выборки менее чем в 74 тысячи наблюдений это невозможно, число вариантов слишком велико (особенно с учётом PTM и химических модификаций). Чтобы сократить число вариантов для соседних остатков введено разделение на 4 группы: 1 – у остатка нет дисооциируемых групп; 2 - у остатка есть протонируемые группы; 3 - у остатка есть диссоциируемые группы; 4 - у остатка есть и протонируемые и диссоциируемые группы. Таким образом, возможно 24 варианта констант для каждого остатка (шкала 24), и хотя для каждого остатка может быть до 3 значений pKa и до 3 pKb, реальное количество переменных в шкале – 1740. Уменьшение числа переменных было получено и за счёт того, что в случае, когда модификация относится к N- или C-концевому остатку и имеет место образование амидной связи (например, ТМТ или iTRAQ), то такая модификация рассматривается как самостоятельный остаток, а аминокислотный остаток атрибутируется при этом как внутренний. Обычно для тестирования выборку делят на две части – обучающую и тестовую, но выборка в 73863 наблюдений и так не слишком велика, особенно для шкалы 24. Поэтому в качестве тестовых рассматривали все пептиды по всем 25 наборам данных, не вошедшие в обучающую выборку. Хотя количество ошибочных идентификаций и неправильно определённых значений pI там больше, но в то же время подавляющая часть пептидов идентифицирована правильно, как и значения pI в существенном числе случаев. Мерой при этом может служить процент предсказаний, совпадающих с определёнными экспериментально. Кроме того, провели сравнение с данными, полученными различными методами, приведёнными в работе [15]. Тестирование качества предсказания для белков проводили на выборках из работ [15, 24]. Данные последней были загружены из World-2DPAGE Repository (https://world-2dpage.expasy.org/repository/) [25]. Оптимизацию значений pKa в шкалах проводили с использованием метода Монте-Карло (до 100000 итераций) с последующей процедурой покоординатного наискорейшего спуска (независимо по каждому из значений pKa). В качестве оптимизируемого параметра использовали среднюю абсолютную ошибку предсказания (MAE). Процедуру по всем значениям pKa повторяли циклически до 100 раз или до прекращения изменений оптимизируемой величины. Расчёт значений pI при оптимизации проводили численно (точность до 0.001 величины pH) с использованием уравнения Хендерсона-Хассельбаха:
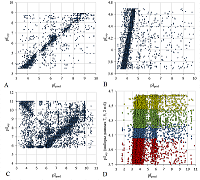
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ В результате оптимизации минимальная величина MAE для шкал 3 и 24 составила 0.128 и 0.123 единицы pH. Качество предсказания значений pI на примере обучающей выборки можно оценить по рисунку 3. Значения R2 обучения приведены на диаграммах в двух вариантах с учётом константы и без. По диаграммам сложно сказать, в чём преимущество шкалы 24, но это видно по распределению АЕ (рис. 3С). На качество предсказания для выбросов (за которые, следуя работе [15], приняты значения AE, превышающие 0.25) детализация шкалы особо не влияет, но для части пептидов c AE в пределах 0.25 величина pI предсказывается точнее (в предел до 0.05 попадает на 10% больше). Больших различий между шкалами быть и не может, так как по сути отличие второй важно только для случаев, когда рядом расположены заряженные остатки, а большинство аминокислотных остатков не имеет заряженных групп. Не для всех из 24 вариантов было достаточно наблюдений по каждому из типов остатков. В таком случае недостающие значения дополняли, используя усреднённое значение для других типов остатков, схожих по спектру диссоциируемых и протонируемых групп. Это важно для предсказания пептидов, не входящих в обучающую выборку.
Результаты тестирования для всех 25 наборов данных представлены в таблице 2.
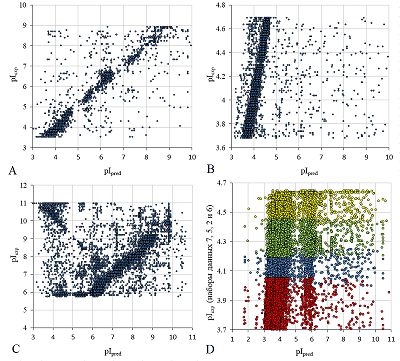
При предсказании для наборов данных 2, 5-7 от 40% до 60% наблюдений попадают в область с АЕ менее 0.25, а на диаграмме, объединяющей все 4 выборки (рис. 4D), можно видеть общую полосу, соответствующую зоне пептидов, позиционированных в соответствии с их pI, и шлейф из пептидов с более основным pI, которые не вышли за пределы стрипа для IEF. Для всех наборов данных с диапазоном от pH 3 до 10 (пример на рисунке 4A) количество выбросов не превышает 32% в худшем случае (напомним, что все они были обеднены «правильными» значениями). Наборы данных 13 и 17 не были выравнены с остальными и при этом имеют достаточно узкий диапазон значений pI. Так как для этих наборов имеется максимум на графике распределения МАЕ в районе 0.2-0.25, весьма вероятно, что имеет место системная ошибка по определению диапазона pH. Выборки 1 и 18 (рис. 4B) содержат пептиды с iTRAQ меткой, причём в обучающую выборку вошло менее 10% от общего числа пептидов, тем не менее они имеют лучший результат предсказания. Отдельно нужно отметить данные по наборам 24 (рис. 4С) и 25. Мы не можем объяснить наличие в них больших групп пептидов, для которых pI предсказывается с точностью до наоборот под углом 90 градусов к основному тренду, ничем иным, кроме как наличием загрязнений. Имея немного представления о практике подобных экспериментов, наличие таких загрязнений можно предположить для набора 25, данные для отдельных нарезок которого представлены в обратном порядке (от большего значения pI к меньшему). Но так как точный порядок масс-спектрометрического анализа проб неизвестен, то сделать конкретный вывод о возможности подобного загрязнения нельзя. В среднем, даже если учитывать выборки 13 и 17, в пределах АЕ до 0.25 единицы pH находятся предсказания для 77% пептидов (если принимать за величину pI для конкретного пептида медианное значение по всем наборам данных).
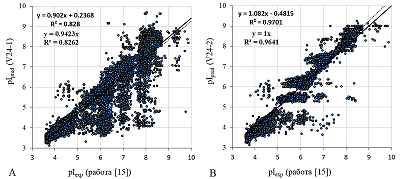
Сравнение качества предсказания по шкале 24 с предсказаниями с использованием шкал других авторов можно провести по данным работы [15], в которой проводится сравнение собственного метода на основе SVM с большим числом вариантов шкал для расчётов по уравнению Хендерсона-Хассельбаха. Все данные предсказаний для выборки в 25% тестовых пептидов, кроме наших, взяты из файлов дополнительных материалов работы [15]. Распределение средней ошибки и R2 представлены в таблице 3. В таблицу вошли только 5 лучших методов, использующих уравнение Хендерсона-Хассельбаха. На первый взгляд наша шкала (колонка V24-1 в таблице), несмотря на формально худшее значение R2 по распределению абсолютной ошибки и количеству выбросов (согласно работе [15] за них принимаются значения, отличающиеся более чем на 0.25 значений pH), даёт результат, сравнимый с методами, использующими уравнение Хендерсона-Хассельбаха, но проигрывает при сравнении с предсказаниями, полученными с использованием SVM. Однако в работе [15] для формирования обучающей и тестовой выборок использовали те же самые наборы данных, которые входят и в число использованных в данной работе [14, 21]. В обеих работах указано, что пробы были обработаны йодацетамидом, а в работе [21] использовали и TMT-метки. Об этих фактах при описании выборки в работе [15] не упоминается. Если заменить все остатки цистеина на карбамидметилцистеин (+57.02 Da), а к пептидам с pIэксп больше 5 (данные из [14] и [21] имеют пересечение в диапазоне от 3 до 4.9, в котором пептиды могут быть как с TMT, так и без) добавить TMT (+229.16), то результат (колонка V24-2) становится сравним с результатами IPC2
Для наглядности изменения предсказываемых значениях pI с и без учёта PTM приведены на рисунке 5. Следует отметить, что используя вышеописанную методику подбора шкалы pKa и обучающую выборку из работы [15], можно получить более хороший результат (колонка V24-3) для предсказания значений pI тестовой выборки. Однако, использовать его для предсказания можно только для пептидов, имеющих облигатные модификации (карбамидметилцистеин и наличие TMT-меток).
Предсказание pI для пептидов – задача достаточно специфическая, значительно чаще необходимо предсказание pI для белков. Результаты предсказания с использование шкалы 24 для двух выборок белков представлены на рисунке 6. Так как, в отличие от работы [15], выборка с белками ни в какой её части не была использована в качестве обучающей, то для получения более полной картины для предсказания использовали всю выборку, а не 25%-ную часть (рис. 6A). В работе [15] для 25% выборки наилучший R2=0.59 (для метода на основе уравнения Хендерсона-Хассельбаха и без обучения на выборке белков 0.52), при этом наименьшее число выбросов (абсолютная ошибка >0.5) – 247 (43%). При использовании нашей шкалы на вчетверо большей выборке R2 = 0.52, а число выбросов –1052 (46%). Данная выборка представляет собой коллекцию данных, полученных разными исследователями; в работе [15] в случае наличия для одного идентифицированного белка нескольких значений pI брали среднее значение. Конкретная протеоформа заведомо не была известна. На рисунке 6 B представлены результаты предсказания для белков Staphylococcus aureus [24], полученные одной группой (в двух экспериментах с разным диапазоном pH). В данном случае и вероятность того, что белки имеют PTM намного меньше, чем у эукариотических клеток. Соответственно, результат предсказания значительно лучше, хотя тот факт, что результаты получены при анализе двух электрофорезов и потом сведены вместе, хорошо виден.
ЗАКЛЮЧЕНИЕ Полученные шкалы могут быть использованы как для предсказания значения pI пептидов с модификациями или без, так и для предсказания pI белков с учётом некоторых PTM и химических модификаций. Для использования данных шкал созданы два варианта программы. Первый (написан на JavaScript) имеет графический интерфейс пользователя (рис. 7), работает как WEB-приложение и доступен по адресу http://pIPredict3.ibmc.msk.ru.
Для данного варианта установлено ограничение по размеру загружаемой выборки – 1000 белков (пептидов). Если встречается запись с модификацией, не имеющейся в шкале, то такой остаток можно проигнорировать, либо считать не модифицированным. Второй вариант – исполняемая программа для win32, написанная на С++, работает из командной строки и не имеет ограничений по количеству белков (пептидов). Программа доступна для скачивания по тому же адресу. В случае «неизвестных» модификаций остаток игнорируется. Описание возможных модификаций и выборок данных, использованных в работе, также доступно в дополнительных материалах. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Данная работа не содержит каких-либо исследований с использованием людей и животных в качестве объектов исследования. ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021-2030 годы). КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. К данной статье приложены дополнительные материалы, свободно доступные (http://dx.doi.org/10.18097/BMCRM00161) на сайте журнала. ЛИТЕРАТУРА
|