|
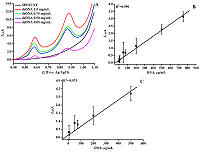
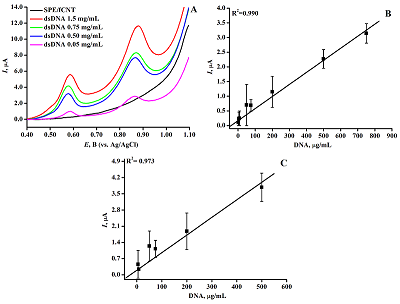
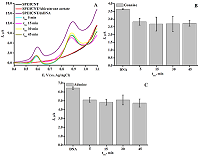
СОДЕРЖАНИЕ СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Рисунок 1Структурная формула абиратерона (17-(3-пиридил)андроста-5,16-диен-3β-ол). Рисунок 3(А) ДИВА ПГЭ/фУНТ с различной концентрацией дцДНК. На рабочую поверхность электродов наносили 60 мкл дцДНК с концентрацией: (-) 1.5 мг/мл, (-) 0.75 мг/мл, (-) 0.5 мг/мл, (-) 0.05 мг/мл, (-) ПГЭ/фУНТ. Электролит 100 мМ калий-фосфатный буфер, содержащий 50 мМ NaCl (pH 7/4) Потенциал электронакопления (аккумулирования) 0/4 В, время аккумулирования 15 минут, амплитуда импульса 0/025 В, шаг потенциала 0/005 В, длительность импульса 50 мс, амплитуда модуляции 0/05 В. Все потенциалы отнесены к электроду сравнения Ag/AgCl. (B) Зависимость величины интенсивности тока электроокисления гуанина от концентрации дцДНК. (C) Зависимость величины интенсивности тока электроокисления аденина от концентрации дцДНК.
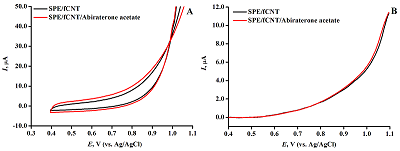
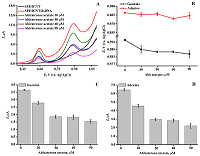
Рисунок 4(А) ЦВА ПГЭ/фУНТ/ацетат абиратерона с концентрацией ацетата абиратерона 10 мМ в диапазоне потенциалов 0.4–1.1 В (отн Ag/AgCl). (B) ДИВА ПГЭ/фУНТ/ ацетат абиратерона в диапазоне потенциалов 0.4–1.1 В (отн Ag/AgCl).
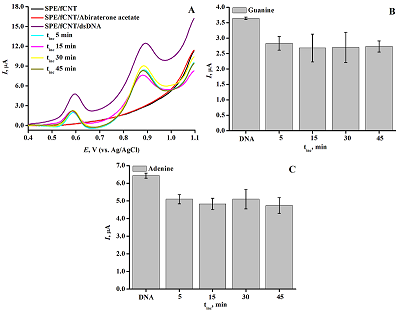
Рисунок 5(А) ДИВА ПГЭ/фУНТ. На рабочую поверхность электрода наносили 60 мкл раствора дцДНК 1.5 мг/мл и раствор дцДНК 1.5 мг/мл с 50 мкМ абиратероном с различным временем инкубации комплекса: (-) 5 мин, (-) 15 мин, (-) 30 мин, (-) 45 мин, (-) 60 мин, (-) ПГЭ/фУНТ, (-) ПГЭ/фУНТ/абиратерон, (-) ПГЭ/фУНТ/ДНК. Параметры эксперимента: диапазон потенциалов 0.4–1.1 В, амплитуда импульса 0.025 В, шаг потенциала 0,005 В, длительность импульса 50 мс, амплитуда модуляции 0.05 В. (B) Зависимость интенсивности сигнала электроокисления гуанина при взаимодействии абиратерона с дцДНК от времени инкубации. (C) Зависимость интенсивности сигнала электроокисления аденина при взаимодействии абиратерона с дцДНК от времени инкубации.
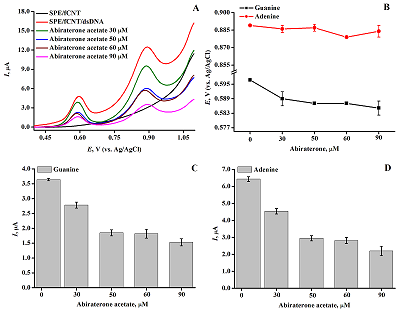
Рисунок 6(А) ДИВА ПГЭ/фУНТ/дцДНК и ПГЭ/фУНТ/дцДНК/ ацетат абиратерона с различной концентрацией препарата: (-) 30 мкМ, (-) 50 мкМ, (-) 60 мкМ, (-) 90 мкМ, (-) ПГЭ/фУНТ/ацетат абиратерона, (-) ПГЭ/фУНТ/дцДНК. Параметры ДИВА: диапазон потенциалов 0.4–1.1 В, амплитуда импульса 0.025 В, шаг потенциала 0.005 В, длительность импульса 50 мс, амплитуда модуляции 0.05 В. (B) Зависимость значений потенциалов электроокисления пуриновых оснований дцДНК от концентрации ацетата абиратерона в диапазоне 0–90 мкМ. (C) Влияние терапевтических концентраций ацетата абиратерона (0–90 мкМ) на интенсивность электроокисления гуанина дцДНК; (Г) Влияние терапевтических концентраций ацетата абиратерона (0–90 мкМ) на интенсивность электроокисления аденина дцДНК.
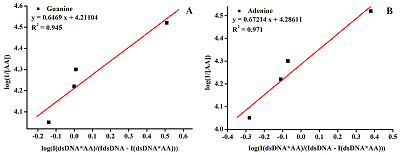
Рисунок 7(А) Зависимость log(1/[АА]) от log[IдцДНК-АА]/(IдцДНК- [IдцДНК-АА]) для определения значения константы связывания для гуанина. (B) Зависимости log(1/[АА]) от log[IдцДНК-АА]/(IдцДНК-[IдцДНК-АА]) для определения значения константы связывания для аденина.
Таблица 1Электроаналитические характеристики дцДНК с использованием ПГЭ/фУНТ.
Таблица 2Электроаналитические характеристики комплекса дцДНК-АА на ПГЭ/фУНТ.
Таблица 3Электрохимический коэффициент токсического эффекта ацетата абиратерона 30÷90 мкМ на дцДНК.
|
Взаимодействие противоопухолевого препарата ацетата абиратерона с дцДНК
1Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121 Москва, ул. Погодинская, 10; *e-mail: viktoria.shumyantseva@ibmc.msk.ru 2Российский национальный исследовательский медицинский университет имени Н.И. Пирогова, Москва Ключевые слова: электроанализ; углеродные нанотрубки; модифицированный электрод; абиратерон; ДНК; константа связывания DOI: 10.18097/BMCRM00174 ВВЕДЕНИЕ Выяснение механизма действия лекарства на ДНК является одним из ключевых вопросов фармакогеномных исследований для понимания природы многих типов заболеваний, механизма действия лекарств на организм и разработки новых фармацевтических препаратов [1-3]. ДНК является фармакологической мишенью многих лекарств, которые, связываясь с ДНК, могут влиять на жизненно важные функции клеток, воздействуя на экспрессию, модифицируя гистоны, вызывая карциногенез или мутагенез. Химическая реакционная способность лекарственных препаратов и образование активных форм кислорода может приводить к повреждению ДНК и разрывам сахарофосфатного остова [4]. Взаимодействие лекарства с двухцепочечной ДНК может происходить различными способами, включая интеркаляцию, связывание в бороздках спирали, электростатическое взаимодействие, расщепление ДНК и встраивание аналогов нуклеозидов [5, 6]. Понимание механизмов комплексообразования ДНК с лекарством имеет решающее значение для создания новых лекарственных препаратов [7]. Индукция повреждения или разрывов цепи ДНК является серьезной проблемой для многих противоопухолевых терапий, мишенью которых является ДНК. Кроме того, для экспериментальной и клинической онкологии необходим дизайн новых ДНК-связывающих молекул, способных изменять архитектуру ДНК и блокировать транскрипцию, не вызывая повреждения ДНК. Количественный анализ ДНК, нуклеотидов, нуклеозидов, гетероциклических оснований (ГЦО) может быть проведен различными методами: абсорбционной спектроскопией, где регистрируется интегрированный спектр поглощения ДНК, флуоресцентной спектроскопией с введением флуоресцентных «меток», полимеразной цепной реакцией (ПЦР) с необходимым набором дополнительных реактивов и автоматизированного оборудования [8, 9]. Электрохимический метод количественного анализа ДНК имеет ряд преимуществ по сравнению с другими методами. Его отличают высокая чувствительность, малый объем проб (0.5-60 мкл), непродолжительное время анализа, регистрация пуриновых и пиримидиновых ГЦО при соответствующем выборе типа рабочих электродов [10-17]. Кроме того, наноструктурирование рабочей поверхности электродов (модификация электродов наноматериалами) позволяет подобрать оптимальные условия, сделать селективный сенсор выбранной электрохимической реакции определяемого вещества, а также обеспечить необходимые аналитические характеристики метода, такие как биосовместимость, предел обнаружения, широкий диапазон определяемых концентраций аналита. Электрохимические ДНК-биосенсоры используются для анализа взаимодействия с ДНК при обнаружении и количественном определении химических веществ, таких как лекарства, метаболиты, биомаркеры [11-14]. Рак предстательной железы является одним из наиболее распространённых онкологических заболеваний. Фармакотерапия этого заболевания направлена на снижение уровня андрогенов за счёт использования ингибиторов цитохрома P450 17А1 (17α-гидроксилазы, 17,20-лиазы) как ключевого фермента биосинтеза андрогенов, а также на блокирование взаимодействия андрогенов с рецепторами андрогенов опухолевых клеток [18-27]. Абиратерон (рис. 1) широко используют для лечения пациентов, страдающих метастазирующим кастрационно-резистентным раком предстательной железы; при его приёме в форме ацетата абиратерона (АА) в дозе 1000 мг/день наблюдается снижение уровня тестостерона до <50 нг/мл, что приводит к сокращению пролиферации опухолевых клеток, чувствительных к тестостерону [24-26]. Несмотря на активное использование абиратерона в медицинской практике, фармакологические мишени этого препарата недостаточно исследованы, за исключением ряда изоформ цитохрома P450 (21A2, 51A1, 11A1, 11B2, 19A1, 3A4, 2D6, 2C8), а также 3β-гидроксистероиддегидрогеназы [19-21]. Взаимодействие абиратерона с ДНК ранее было исследовано методами флуоресцентной и абсорбционной спектроскопии [26]. На основе анализа спектральных характеристик и величины изменения свободной энергии ΔG был сделан вывод о термодинамически выгодном самопроизвольном процессе, включающем механизм образования водородных связей и Ван-дер-Ваальсовых взаимодействий за счет связывания абиратерона с ДНК в малой бороздке.
Цель данной работы – исследование механизма взаимодействия АА с дцДНК электрохимическими методами и оценка диапазона концентраций, оказывающих влияние на электрохимический сигнал окисления пуриновых азотистых оснований, входящих в состав дцДНК. МЕТОДИКА Трехконтактные печатные графитовые электроды (ПГЭ) были приобретены у «ColorElectronics» (Россия). Электрохимические измерения выполняли на приборе потенциостат/гальваност Autolab 302N («Metrohm Autolab BV», Нидерланды) с программным обеспечением Nova (версия 2.0). Все электрохимические потенциалы приведены относительно хлоридсеребряного (Ag/AgCl) электрода сравнения. Функционализированные углеродные нанотрубки (фУНТ) были приготовлены согласно методике [28]. Двухцепочечная ДНК, выделенная из молок осетровых рыб, и плазмида pBR322 были получены от «Sigma-Aldrich» (США). АА был получен от «Chemleader Biomedical» (Китай). Все остальные реактивы отечественного производства имели аналитическую чистоту и использовались без дополнительной очистки. Водные растворы были приготовлены с использованием воды Milli-Q, очищенной с помощью системы очистки Milli-Q water («Millipore», США). Все электрохимические измерения проводили при комнатной температуре в 100 мМ калий-фосфатном буфере (рН 7.4) с 50 мМ NaCl в качестве фонового электролита. Прямое электрохимическое окисление дцДНК регистрировали при помощи метода дифференциально-импульсной вольтамперометрии (ДИВА), используя следующие параметры: диапазон потенциалов 0.4–1.1 В, амплитуда импульса 0.025 В, шаг потенциала 0.005 В, длительность импульса 50 мс, амплитуда модуляции 0.05 В. Исходный раствор дцДНК (3 мг/мл) готовили в 100 мМ калий-фосфатном буфере (pH 7.4) с 50 мМ NaCl. Разведение исходного раствора дцДНК проводили тем же буфером. Исходный 10 мМ раствор АА готовили в смеси этанола и 100 мМ калий-фосфатного буфера, 50 мМ NaCl (pH 7.4) в соотношении 1:1. Разведение исходного раствора АА проводили тем же буфером. Для электрофореза готовили 1% агарозный гель, затем его нагревали и добавляли бромистый этидий до полного остывания, затем планшет помещали в ванну с трис-ацетатным буфером (TAE). Рабочую поверхность ПГЭ модифицировали 2 мкл дисперсией 1 мг/мл фУНТ в этаноле, приготовленной при помощи ультразвуковой дезинтеграции в течение 5 мин. фУНТ исследовали методом сканирующей электронной микроскопии (СЭМ) на приборе HitachiS-5500 («Hitachi», Япония). Внешний диаметр фУНТ составлял 56 ± 10 нм. Электронакопление дцДНК (из 60 мкл раствора дцДНК заданной концентрации) на поверхности модифицированного электрода ПГЭ/фУНТ проводили при потенциале 0.4 В в течение 15 мин. Во всех экспериментах использовали горизонтальный режим измерений. Для исследования взаимодействия дцДНК-АА комплекс дцДНК с АА готовили в заданных концентрациях и инкубировали в течение 15 мин, затем проводили измерения сигнала окисления пуриновых азотистых оснований дцДНК методом ДИВА. Расчёт электроактивной площади электродов, модифицированных фУНТ, проводили методом циклической вольтамперометрии (ЦВА) в диапазоне потенциалов от 0.8 до -0.3 В, скорости развертки потенциала от 10 до 100 мВ/с. Измерения проводили в вертикальном режиме, электрод погружали в кювету, содержащую 1 мл 5 мМ раствора феррицианида калия в 0.1 М KCl. Расчёт электроактивной поверхности осуществляли из линейной зависимости Ip от корня квадратного скорости развертки потенциала в соответствии с уравнением Рэндлса-Шевчика (1) [29, 30]:
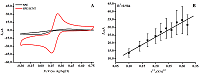
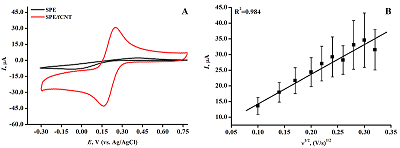
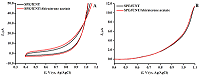
где Ip – ток пика восстановления феррицианида (A), A – площадь электроактивной поверхности электрода (см2), D – коэффициент диффузии (см2/с), ν – скорость развертки потенциала (В/с), C – концентрация феррицианида (М), n – число электронов, участвующих в электрохимической реакции. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Электроаналитические характеристики ПГЭ/фУНТ Для исследования электроаналитических характеристик электродов, модифицированных фУНТ (ПГЭ/фУНТ), были проанализированы циклические вольтамперограммы (ЦВА) с использованием внешнего электролита феррицианида калия K3Fe(CN)6. Методом ЦВА регистрировали сравнительные значения интенсивности сигналов электровосстановления/электроокисления Fe(CN)63-/4- на ПГЭ и модифициированном ПГЭ/фУНТ (рис. 2А). Для расчёта площади электроактивной поверхности была построена зависимость интенсивности электровосстановления от корня квадратного из скорости сканирования (рис. 2Б).
На ЦВА присутствуют два пика, характеризующие процессы восстановления и окисления на поверхности ПГЭ феррицианида (Fe(CN)63-/4-). Потенциалы восстановления и окисления феррицианида имеют значения 0.162±0.006 В и 0.252±0.003 В (отн. Ag/AgCl) соответственно. Наблюдается линейная зависимость между величиной пика восстановления и корнем квадратным из скорости развертки потенциала, что свидетельствует о диффузионно-контролируемом процессе [29, 31]. Электроаналитические характеристики электродов, модифицированных фУНТ, продемонстрировали существенное увеличение электроактивной площади модифицированных электродов (в 5 раз) по сравнению с немодифицированными электродами (0.0024 см2 и 0.0121 см2 для ПГЭ и ПГЭ/фУНТ соответственно). Количественный анализ дцДНК методом дифференциально-импульсной вольтамперометрии, сопряженной с инверсионной вольтамперометрией Количественный анализ дцДНК проводили на основе анализа интенсивности электрохимического окисления пуриновых гетероциклических оснований аденина и гуанина [32-34]. Измерения проводили методом ДИВА, сопряженной с электронакоплением образца методом инверсионной вольтамперометрии (ИВА) при потенциале 0.4 В в течение 15 мин. Снижение интенсивности пиков электрохимического окисления пуриновых оснований при повторных измерениях показывает необратимость электрохимического процесса (дополнительные материалы). ДИВА системы ПГЭ/фУНТ/ДНК демонстрируют два пика в области потенциалов 0.59 ± 0.03 В и 0.86 ± 0.04 В (отн. Ag/AgCl), которые соответствуют электрохимическому окислению гуанина и аденина (рис. 3А). Зависимость амплитуды тока электроокисления гуанина и аденина дцДНК от концентрации линейна в диапазоне концентраций 0-750 мкг/мл для гуанина и 0-500 мкг/мл для аденина (рис. 3 Б, В).
Электроаналитические характеристики пуриновых оснований гуанина и аденина приведены в таблице 1. Количественная регистрация дцДНК на ПГЭ/фУНТ является воспроизводимой, а аналитические характеристики ПГЭ/фУНТ/ДНК сравнимы с опубликованными данными модифицированных электродов, которые использовались для регистрации дцДНК ранее (дополнительные материалы). Электроаналитические характеристики демонстрируют низкий предел определяемых концентраций дцДНК и высокую чувствительность анализа.
Модификация электродов функционализированными углеродными нанотрубками (ПГЭ/фУНТ) позволяет регистрировать ДНК на основе интенсивности электрохимического окисления гуанина и аденина при потенциалах 0.59 ± 0.03 В и 0.86 ± 0.04 В (отн. Ag/AgCl) соответственно, что может быть использовано для электрохимического анализа комплекса дцДНК-АА. Амплитуды токов электрохимического окисления пуриновых оснований (10-6 мкА для концентраций ДНК 0.2–3.0 мг/мл) позволяют чётко и достоверно проследить влияние препарата на биомолекулу. Исследование электроактивности ацетата абиратерона на ПГЭ/фУНТАнализ электрохимической активности АА выполнен с использованием графитовых электродов с модификацией рабочей поверхности фУНТ. Электроактивность 10 мМ АА исследовали методами ЦВА и ДИВА в 60 мкл буфера в диапазоне потенциалов 0.4 –1.1 В (отн Ag/AgCl), который соответствует диапазону значений потенциалов регистрации дцДНК (рис. 4).
АА не проявляет электроактивность в области значений потенциалов электроокисления пуриновых оснований с использованием ПГЭ/фУНТ, что позволяет проводить электрохимический анализ комплекса дцДНК-АА на основе регистрации сигналов электрохимического окисления гуанина и аденина. В отличие от исследований, проведенных спектральными методами при постоянной концентрации АА и варьируемой концентрации дцДНК [26], алгоритм анализа данного исследования заключался в исследовании влиянии возрастающих концентраций АА на интенсивность сигналов электрохимического окисления гуанина и аденина при постоянной концентрации дцДНК (1.5 мг/мл). Такой подход дает информацию о концентрационной зависимости противоопухолевого препарата на генетический материал и позволяет сделать выводы о влиянии препарата на ДНК. Оптимизация условий регистрации комплекса дцДНК*АА Выбор оптимальных условий для регистрации влияния препарата на дцДНК основывался на определении наибольшего влияния на интенсивность электроокисления дцДНК (1.5мг/мл) и АА (50 мкМ) после инкубации методом ДИВА (рис. 5). Пик в области потенциалов 0.59 ± 0.03 В соответствует электроокислению гуанина, пик в области потенциалов 0.86 ± 0.04 В соответствует электроокислению аденина.
Как следует из рисунка 5 Б, В, наиболее интенсивное уменьшение тока электроокисления дцДНК наблюдается для времени инкубации 15 мин, это время было выбрано оптимальным для анализа образования комплекса дцДНК-АА. Исследование влияния концентрации ацетата абиратерона на комплексообразование с ДНКТерапевтические концентрации лекарственных препаратов в крови пациентов регистрируются в диапазоне микромолярных значений [25-27]. Для анализа влияния АА на ДНК 0–90 мкМ препарата инкубировали с дцДНК в течение 15 минут и регистрировали ДИВА в диапазоне потенциалов 0.4–1.1 В. На вольтамперограмме наблюдаются два пика в области потенциалов 0.6 В и 0.9 В, что соответствует электроокислению комплекса дцДНК-АА для гуанина и аденина соответственно. Согласно данным таблицы 2 были построены гистограммы зависимости интенсивности электроокисления гуанина и аденина от концентрации АА (рис. 6).
В таблице 2 представлены электроаналитические характеристики комплексов электродов дцДНК-АА. Как следует из рисунка 6 А, В, Г, регистрируется концентрационно-зависимое (в диапазоне концентраций 0–90 мкМ) снижение интенсивности электроокисления гуанина и аденина дцДНК.
При взаимодействии лигандов с ДНК по механизму интеркаляции регистрируется смещение потенциалов электрохимического окисления ГЦО в положительную (анодную) область потенциалов, смещение в отрицательную (катодную) область потенциалов может свидетельствовать об образовании водородных связей и/или электростатических взаимодействиях в системе ДНК-лиганд [1, 10, 35]. Зависимость потенциала электроокисления от концентрации АА в диапазоне 0–90 мкМ демонстрирует смещение в катодную область на 12 мВ для гуанина и на 5 мВ для аденина, что отражает более термодинамически выгодный электрохимический процесс (рис. 6Б). Смещение потенциала в отрицательную область свидетельствует об отсутствии интеркаляционных взаимодействий и о взаимодействии с бороздками ДНК за счет образования водородных связей [1, 10, 35]. В процессе взаимодействия образуется электрохимически менее активный, чем дцДНК, комплекс дцДНК*АА, что отражается в снижении амплитуд электрохимического окисления гуанина и аденина. Результаты электрохимического анализа взаимодействий находятся в соответствии с ранее проведенными спектральными исследованиями комплекса ДНК/абиратерон [26]. Электрохимический коэффициент токсичности препарата (S%) оценивали при каждой концентрации АА как величину соотношения тока электроокисления гуанина или аденина по формуле (2):
где Sb и Ss – интенсивности токов электроокисления пуриновых оснований до и после взаимодействия АА с дцДНК соответственно. По принятым критериям препарат считается нетоксичным, если коэффициент токсичности (S) больше 85%; препарат относится к умеренно токсичным, если S имеет значения от 50 до 85%, и препарат токсичен, если S меньше 50% [17, 31, 33, 34]. Электрохимические коэффициенты токсического эффекта для концентраций препарата 30 мкМ, 50 мкМ, 60 мкМ и 90 мкМ, рассчитанные по снижению интенсивности амплитуд электроокисления гуанина и аденина, приведены в таблице 3.
При концентрациях АА выше 60 мкМ происходит интенсивное снижение сигналов ДНК (величина S меньше 50%), что позволяет отнести эти концентрации к токсичным. Терапевтически значимые концентрации АА 20–360 нМ для стандартной дозы 1000 мг/день [25-27, 35, 36] существенно ниже, что позволяет предположить отсутствие токсичности для более низких концентраций этого препарата. Определение константы связывания Константу связывания для процесса образования комплекса дцДНК-АА (3) рассчитывали по уравнению (4) при постоянной концентрации дцДНК 1.5 мг/мл.
где Kb - константа связывания, (М-1), IдцДНК – максимальная амплитуда тока электроокисления дцДНК, IдцДНК-АА – максимальная амплитуда тока электроокисления комплекса дцДНК-АА [1, 26, 37]. Пересечение линейного графика зависимости log(1/[АА]) от log([IдцДНК-АА])/(IдцДНК - [IдцДНК-АА]) использовали для определения значения константы связывания гуанина и аденина (рис. 7).
Для гуанина значение константы связывания равно 1.63х104 М-1 и 0.96, а для аденина 1.93х104 М-1. Константы связывания для интеркаляции и взаимодействия лиганда с бороздками ДНК находятся в пределах 105–109 М-1. Для электростатических взаимодействий константы связывания имеют существенно более низкие значения (как правило, 103 М-1). На основании анализа полученных результатов по смещению потенциалов электрохимического окисления гуанина и аденина при образовании комплекса ДНК-лекарство и значений констант связывания можно сделать вывод о механизме взаимодействия АА с бороздкой дцДНК за счет образования водородных связей, способствующих таким взаимодействиям [17, 26, 37-41]. Полученные нами результаты находятся в соответствии с результатами, полученными с помощью спектральных исследований и молекулярного моделирования комплекса дцДНК-абиратерон, исключающими интеркаляционные взаимодействия [26]. Исследование влияния ацетата абиратерона на длину фрагментов дцДНК и плазмиды Многие лекарственные препараты способны вызывать фрагментацию ДНК, что может приводить к нежелательным побочным эффектам. Для исследования фрагментации и целостности дцДНК после взаимодействия с АА в диапазоне концентраций 0–50 мкМ были проведены исследования методом электрофореза в агарозном геле. Объектом исследования была также плазмида pBR322. Электрофореграмма подтвердила целостность молекул ДНК и плазмиды pBR322 после взаимодействия с АА: не наблюдалось фрагментов с меньшей молярной массой, чем исходные мишени (дополнительные материалы). ЗАКЛЮЧЕНИЕ Разработан чувствительный электрохимический метод анализа ДНК с пределом определяемых концентраций 1-1.6 мкг/мл. ДНК-сенсор был применен для исследования взаимодействия противоопухолевого препарата АА с дцДНК при одновременной регистрации изменений токов электроокисления гуанина и аденина, что является преимуществом по сравнению со спектральными методами, где регистрируется интегральный сигнал ДНК. При концентрациях выше 60 мкМ АА влияет на дцДНК, вызывая снижение сигналов ДНК более, чем на 50%. Смещение потенциалов электроокисления аденина и гуанина ДНК в катодную область при увеличении концентрации АА отражает механизм взаимодействия препарата с бороздкой дцДНК за счет образования водородных связей и исключает интеркаляцию лекарства. АА не вызывает фрагментацию ДНК, что подтверждено отсутствием фрагментов ДНК с меньшей молярной массой при анализе электрофореграмм. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов. ФИНАНСИРОВАНИЕ Работа выполнена в рамах Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период 2021 - 2030 годы (№122030100168-2). КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|