|
Идентификация протеоформ в результатах 2D электрофореза по предсказанным значениям величины изоэлектрической точки Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121, Москва, ул. Погодинская, 10; e-mail: aleona.rybina@ibmc.msk.ru Ключевые слова: изоэлектрическая точка; 2D электрофорез; анализ протеформ DOI: 10.18097/BMCRM00191 ВВЕДЕНИЕ
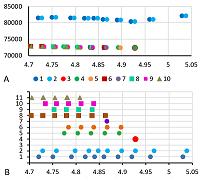
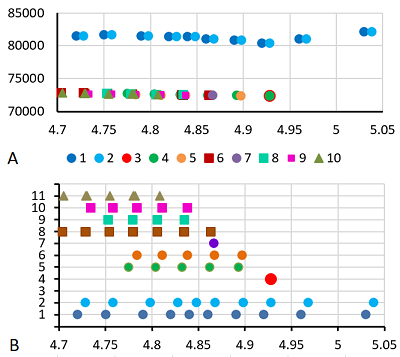
Посттрансляционные модификации (ПТМ) белков, в частности химические модификации боковых радикалов аминокислотных остатков [1], играют важную роль в регуляции многих биохимических процессов в клетке в норме и при патологии. Однако идентификация белков с ПТМ часто не является основной задачей исследования и поэтому методически не поддерживается. Между тем, в протеомных масс-спектрометрических исследованиях, наряду с собственно идентификацией характеристических пептидов (а, следовательно, и белков), можно идентифицировать и их модифицированные формы [2, 3]. Но это, как правило, требует специфических настроек аналитического программного обеспечения. Анализ данных двумерного (2D) гель-электрофореза [4] далеко не всегда позволяет выявить ПТМ. При детекции белков методом иммуноферментного анализа не удается различить модифицированные и немодифицированные формы белков. При проведении MS/MS анализа исследователю нет необходимости получить полное покрытие белка; как правило, достаточно идентифицировать 2-3 характеристических пептида. В связи с этим исследователи редко настраивают программы идентификации на поиск широкого спектра ПТМ, особенно учитывая тот факт, что нередко вторичные спектры, потенциально содержащие такие ПТМ пептидов, менее качественные. В то же время с высокой долей вероятности масса первичных ионов для пептидов, содержащих модификации, измерена и записана. Иногда может существовать и информация по фрагментации таких пептидов, но нужно применить другое программное обеспечение, либо сменить настройки при обработке исходных данных [5]. В данной работе рассмотрена возможность генерации гипотез о конкретных ПТМ белков на карте 2D гель-электрофореза на основании предсказания величины изоэлектрической точки белка (pI). Само по себе подобное предсказание мало что доказывает, но с помощью имеющейся совокупности косвенных данных (о наличии первичного иона неидентифицированного пептида, совпадающего по массе с предсказанным пептидом с ПТМ, совпадении экспериментально определённого для того же пептида и предсказанного для пептида с ПТМ времени удержания) вероятность правильной идентификации конкретных протеоформ существенно повышается. Список подобных косвенных данных можно расширить, добавив определённую величину pI для пептидов в процедуре изоэлектрического фокусирования пептидов, изменение молекулярного веса (MW) белков при модификации и т.д. Ранее мы представили программу pIPredict 3 для предсказания изоэлектрической точки (pI) пептидов и белков [6], основанную на использовании уравнения Хендерсона-Хассельбаха [7]. При этом табличные значения pKa были рассчитаны на основании данных, полученных для большой выборки пептидов, в том числе и с различными химическими и посттрансляционными модификациями, и программа может предсказывать pI для белков с ПТМ. Существуют и другие программы, позволяющие делать подобные предсказания [8, 9], но отличие от них спектр ПТМ в pIPredict 3 охватывает большую часть возможных в физиологических условиях. Важно, что средняя ошибка предсказания pIPredict 3 для пептидов была меньше 0.1 единицы pH. Возможности по предсказанию конкретных ПТМ у идентифицированных белков продемонстрированы на конкретных характерных примерах из реальных карт 2D гель-электрофореза. МЕТОДИКА Источники данных и стандартизация значений pI В качестве демонстрационных примеров для предсказания протеоформ белков с известными значениями pI были использованы несколько наборов данных, полученных в экспериментах по 2D гель-электрофорезу, экстрагированные как из оригинальных статей, так и депонированные в World-2DPAGE Repository [10]. Карта 1 [11] Целью работы [11] был поиск маркеров опухолей в плазме крови человека для создания профилей биомаркеров опухоли (в том числе с ПТМ). Авторы постулировали, что с помощью классического, секционного и полувиртуального 2D электрофореза в сочетании с тандемной масс-спектрометрией с жидкостной хроматографией и ионизацией электрораспылением (LC ESI-MS/MS) они создали паттерны 2D электрофореза для наиболее распространенных белков плазмы (более 100 наборов). Это единственная работа из рассмотренных статей, в которой обсуждаются ПТМ. Однако в основном обсуждаются данные из Uniprot [12], за исключением окисления метионина, фосфорилирования серина, треонина и тирозина, ацетилирования лизина. Упомянутые модификации в работе искали при анализе масс-спектрометрических данных. Конкретные аминокислотные остатки, для которых были выявлены ПТМ, в материалах статьи не указаны. Карта 2 [13] В данной работе при помощи двумерного разностного гель-электрофореза (2D-DIGE) исследователи сравнили белковые карты биоптатов желудка пациентов с аутоиммунным атрофическим гастритом и контрольной группы. Различающиеся по интенсивности пятна были идентифицированы с помощью LC–MS/MS. Было найдено 67 пятен для 53 белков. Карта 3 [14] В работе исследовали изменение уровня экспрессии белков в шести органах мыши, вызванное 2-дневным голоданием. 2D-DIGE использовали в качестве альтернативного средства для изучения системных адаптивных ответов. В двух органах (мозг и тестикулы) сильных изменений не нашли, для них фотографии гелей не приведены. Фотографии есть для печени, почки, селезёнки и тимуса. Предполагаемые протеоформы есть только на 2D карте печени. При этом доступна только объединённая карта (мыши до и после голодания). Карты из базы данных WORLD-2D PAGE [10] Данные карты аннотированы как референсные и сделаны специально для этого ресурса. Карта 4 (идентификатор в БД – RAT_INS1E_4-7) [15] – клеточная линия INS-1E бета клеток поджелудочной железы крысы. Карта 5 (PLASMA_HUMAN) сделана для плазмы крови человека [16]. Карта 6 (CSF_HUMAN) – спинномозговая жидкость человека [17]. Для трёх последних карт данные были хорошо аннотированы, но не стандартизированы. Подробно проблемы стандартизации данных, полученных из карт 2D гель-электрофореза, рассмотрены в нашей предыдущей работе [18]. Здесь нужно отметить следующее. Так как карты могли быть опубликованы достаточно давно, то в текущих версиях баз данных не всегда можно найти последовательность белка по имеющемуся идентификатору. Не все карты хорошо аннотированы, например, значения pI может понадобиться вычислять по координатам пятна на фотографии, при этом шкала pH может отсутствовать. Часто при аннотации карты всем протеоформам приписывают одно и то же значение pI, полученное расчётным способом. Для серии протеоформ конкретного белка неизвестно, какое из значений соответствует немодифицированной форме. Из общих соображений это может быть самое большое по площади пятно на 2D карте. Другой вариант – выбрать самое оснóвное значение pI, так как большинство физиологических ПТМ смещают значение pI в кислую сторону. Если процедура предсказания pI учитывает различия в положении аминокислотных остатков в последовательности, то в зависимости от объекта исследований может идти речь об удалении N-концевого остатка метионина (в данной работе его удаляли из последовательности перед вычислениями). Достаточно сложно учесть флуктуации электрофоретического сдвига, пропорционального молекулярному весу белков (MW). Этот параметр также зависит от множества факторов: тех же PTM, гликозилирования (на величину pI практически не влияет), наличия в пробах редуцированных форм или фрагментов белков, наличия в составе белков аминокислотных паттернов, характерных для устойчивых элементов вторичной структуры и не денатурирующих полностью [19], различия в способности конкретных белков связывать молекулы SDS. В данной работе при наличии существенных (более 20% от MW белка) отклонений данные отбрасывались. Источники информации о существующих ПТМ и предсказание значений pI За частичным исключением карты 1, для которой данные о возможных ПТМ обсуждаются в работе [11], все данные были получены из БД Uniprot (раздел PTM/Processing) [12]. Предсказание величины pI выполняли с помощью программы pIPredict 3 [6], использовали вариант с учётом N- и C-концевых аминокислотных остатков, но без учёта соседних аминокислотных остатков. Учитывали только ПТМ, для которых программа pIPredict 3 могла рассчитать pI. При наличии нескольких вариантов ПТМ либо нескольких сайтов модификаций формировали варианты последовательностей с различными комбинациями и эти предсказания также рассматривали. Так как при анализе величину изменения MW белка фактически не учитывали, то данные для наглядности представления преобразовывали, как показано на рисунке 1. На всех иллюстрациях данные по значениям pI, полученные из 2D карт, представлены как в виде исходных данных, так и с корректирующей поправкой, позволяющей совместить предполагаемый вариант без ПТМ с виртуальным. В основе данного действия лежит допущение, что: (i) предсказание величины pI для целого белка менее точно, чем предсказание изменения данной величины при минимальных изменения последовательности; (ii) идентифицированные белки могут содержать единичные аминокислотные замены, смещающие общее значение pI в ту или иную сторону. Также выравнивание относительно одного из конкретных значений может определятся природой рассматриваемого белка (пояснения есть в разделе РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ). В ряде случаев смещение выполняли таким образом, чтобы δpI между всеми наблюдаемыми значениями совпадали с δpI отдельного паттерна среди предсказанных значений.
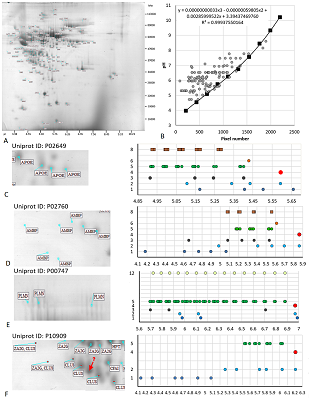
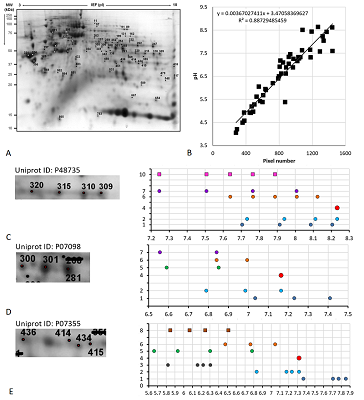
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Для анализа карты 1 (рис. 2A) был использован простой вариант стандартизации величин pI. Так как в статье [11] имеется явным образом указанная шкала pH, то данные пересчитывали напрямую из координат идентифицированных точек с использованием уравнения многочлена третьего порядка (рис. 2B), построенного по координатам меток шкалы pH на фотографии. Для сравнения величин pI, предсказанных программой pIPredict 3 c экспериментально определёнными данными, в качестве предполагаемой немодифицированной формы белка использовали самые оснóвные в серии точек, идентифицированных как один белок. В качестве примеров рассматривали серии не менее чем из 3 точек (протеоформ).
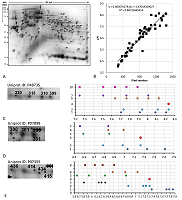
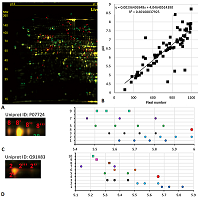
Белок аполипопротеин E (Uniprot ID P02649, MW 36 кДа, рис. 2С). В статье [11] идёт речь о 9 сайтах фосфорилирования, 1 сайте ацетилированиия, 5 сайтах убиквитинирования, 6 сайтах гликозилирования. Так как убиквитинирование добавляет к MW белка 8.6 кДа, то эту ПТМ можно было бы предположить по сдвигу по массе, который не наблюдается. К тому же, программа pIPredict 3 по умолчанию не работает со сшитыми белками, хотя вручную с её использованием приблизительный расчёт pI для такой системы произвести можно, создав химерную последовательность. Гликозилирование не должно существенно менять величину pI, по крайней мере не напрямую (так как нет модификации диссоциирующих групп). Таким образом, для этих модификаций никаких выводов сделать нельзя. pI предполагаемой немодифицированной формы (линия 1, точка справа) отличается от расчётного значения (линия 4) на 0.07 единицы значений pH (т.е. лежит в пределах ошибки предсказания). Если рассмотреть вариант совмещения (линия 2), то наиболее вероятным будет ряд: немодифицированная форма, одиночное ацетилирование (или одиночное фосфорилирование), неидентифицированный вариант и последняя точка – либо тройное или четвертное фосфорилирование, либо комбинация ацетилирования и двойного фосфорилирования. Несложно заметить, что имеется и другой вариант совмещения (линия 3), который даёт свой ряд вариантов: одиночное, двойное, тройное и шестикратное фосфорилирование. Неоднозначность вариантов предсказания pI для фосфорилирования (линия 5) у данной карты определена тем, что в статье не указывается, какие из остатков модифицируются (серин, треонин или тирозин). Белок альфа-1-микроглобулин (P02760, 39 кДа, рис. 2D). Из данных Uniprot следует, что первые 19 аминокислотных остатков отщепляются при процессинге, так что ожидаемый MW 37 кДа (позиционирование на карте 28-33 кДа). В статье упоминается 3 сайта фосфорилирования, 1 ацетилированиия и множество сайтов гликозилирования. Две (возможно три) из семи идентифицированных протеоформ имеют MW на 3-4 кДа меньше остальных, возможно, в результате деградации белка. Предполагаемая немодифицированная форма сильно отличается по величине pI от предсказанного значения. Однако при выравнивании можно предположить наличие двух вариантов рядов для первых 4 наблюдений: (i) немодифицированная форма, неидентифицированная форма, одиночное и двойное фосфорилированиие (либо для последних двух возможно дополнительное ацетилирование); (ii) ацетилирование в каждой их 4 форм и последовательно 1, 2, 3 и 4 фосфорилирования. Белок плазмин (P00747, 90.5 кДа, рис. 2E). В статье речь идёт о 15 возможных сайтах фосфорилирования и большом числе сайтов гликозилирования. Учитывая неоднозначность предсказания pI при фосфорилировании для данного случая в строке 6 приведены усреднённые значения для кратных чисел фосфорилированных остатков от 1 до 15. Наиболее вероятный ряд: немодифицированный белок, 10-кратное и 12-кратное фосфорилирование. В то же время, если внимательно присмотреться к фотографии, можно увидеть затемнения, которые вполне могли бы соответствовать промежуточным вариантам фосфорилирования. А с учётом наличия не менее двух затемнений справа от варианта, принятого за немодифицированный белок, нельзя исключить и другой вариант – тройное, 13- и 15-кратное фосфорилирование. Белок кластерин (P10909, 52.5 кДа, рис. 2F). В статье упоминается о 4 сайтах фосфорилирования и большом числе сайтов гликозилирования. На фотографии одна из точек явным образом выпадает из ряда увеличения MW. Для неё и следующей в кислую сторону точки соответствия не обнаружено. Ряд предсказания: немодифицированная форма, 1, 2 и 3-кратное фосфорилирование. В отличие от карты 1, для карты 2 (рис. 3A) данных о шкале pH в статье нет. Поэтому для определения и стандартизации значений pI был применён другой вариант, а именно построение корреляционного уравнения, связывающего координаты пикселей со значениями pI, предсказанными для идентифицированных белков (рис. 3B). При наличии протеформ использовали значение pI только для самого оснóвного варианта.
Белок митохондриальная изоцитратдегидрогеназа (P48735, 51 кДа, рис. 3C). По данным из Uniprot имеется до 8 сайтов сукцинилирования (рассмотрено до 4), до 21 сайта ацетилирования (рассмотрено до 4). Ряд предсказания: немодифицированная форма, ацетилирование, тройное ацетилирование (либо сукцинилирование и ацетилирование), четверное ацетилирование (либо двойное сукцинилирование, либо сукцинилирование и двойное ацетилирование). Белок желудочная триацилглицероллипаза (P07098, 45 кДа, рис. 3D). В Uniprot есть данные только о сайтах гликозилирования. Однако, используя наиболее часто встречающиеся ПТМ, можно предположить следующий ряд: немодифицированный белок, ацетилирование, фосфорилирование (либо сукцинилирование, либо двойное ацетилирование). Белок аннексин А2 (P07355, 38.5 кДа, рис. 3E). По данным из Uniprot имеется 4 сайта фосфорилирования и 4 сайта ацетилирования. Это пример условно «неудачного» предсказания, так как можно предположить положение немодифицированного белка и точки, соответствующей либо фосфорилированию, либо двойному ацетилированию. Для идентификации ещё двух точек нужно использовать другие ПТМ, данных о которых в Uniprot нет. Для анализа карты 3 (рис. 4A) использовали вариант построения корреляционного уравнения, аналогично карте 2 (рис. 4B). Белок альбумин (P07724, 68.5 кДа, рис. 4C). Наиболее вероятный вариант: немодифицированный белок, нет соответствия, фосфорилирование (либо менее вероятное сукцинилирование) и двойное фосфорилирование. Белок S-аденозилметионин синтаза первого типа (Q91X83, 43.5 кДа, рис. 4D). Логично предположить: немодифицированный белок, нет соответствия и ацетилирование. Возможен и такой вариант: фосфорилирование, сукцинилирование и двойное фосфорилирование.
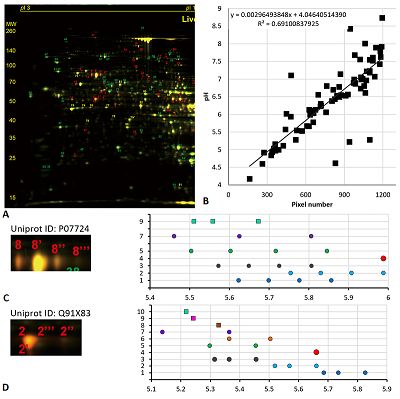
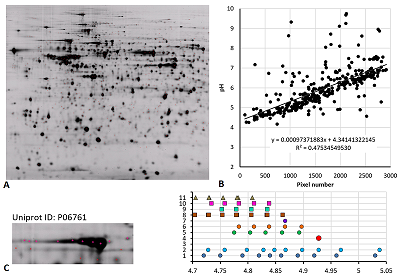
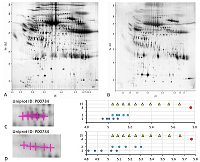
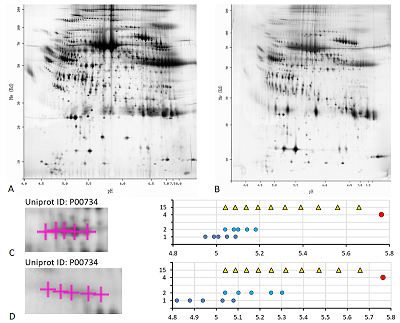
Рассмотрим далее два особых случая анализа карт 2D электрофореза. Первый из них на примере белка шаперона эндоплазматического ретикулума (P06761, 72 кДа) карты 4 (рис. 5A) вариант выбора немодифицированной формы, отличный от ранее использованного. Так как карта 4 хорошо аннотирована, нет нужды перевычислять экспериментальные значения pI (на рисунке 5B продемонстрировано сравнение координат в пикселях по оси pH с предсказанными значениями pI при условии, что для разных протеоформ одного белка эта величина одинаковая). В данном случае (рис. 5C) более адекватный набор предсказаний получен, когда в качестве немодифицированной формы приняли точку с самой большой площадью пятна. Однако, в таком случае для двух более оснóвных точек в Uniprot не описаны ПТМ, которые могли бы дать такой результат. Ещё один пример – это демонстрация важности для исследователя знания особенности белков, которые являются объектом исследования. В данном случае имеется в виду протромбин человека (P00734, 70 кДа) из карт 5 (рис. 6A) и 6 (рис. 6B). Если следовать логике, использованной ранее, то мы должны были совместить самую оснóвную точку в серии с точкой, предсказанной для немодифицированной формы. Но в норме на N-концевом фрагменте белка находится 10 сайтов карбоксилирования, и благодаря специфическому ферменту карбоксилазе [20] все 10 остатков глутамата модифицированы. Таким образом, в данном случае выравнивание нужно проводить по самому кислому значению pI в серии с точкой максимально модифицированного (с точки зрения карбоксилирования) белка. В таком случае протеоформы будут представлять собой варианты белка с неполным карбоксилированием.
ЗАКЛЮЧЕНИЕ Таким образом, использование предсказания величины pI для белков с гипотетическими ПТМ может сформировать набор гипотез, какие конкретно протеоформы наблюдаются на картах 2D электрофореза. При этом важно учитывать всю доступную информацию о конкретном белке: возможные (желательно доказанные) сайты модификации, особенности процессинга, вариабельность аминокислотного состава и т.д. Важно помнить, что сделанные предсказания не доказывают, что наблюдается именно данная протеоформа. Эти гипотезы только указывают направление, в котором нужно искать подтверждающие гипотезу факты. Например, наличие в данных LC-MS/MS первичных ионов с соответствующей массой, со временем ожидания, совпадающим с предсказанным для пептида, который может появиться, если подобная модификация белка имеет место. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Данная работа не содержит каких-либо исследований с использованием людей и животных в качестве объектов исследования. ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021 - 2030 годы) (№ 122030100170-5). КОНФЛИКТ ИНТЕРЕСОВ Автор заявляет об отсутствии конфликта интересов. ЛИТЕРАТУРА
|