|
The Use of Primers Heavily Labeled with Fluorescein in Polymerase Chain Reaction
1Institute of Biomedical Chemistry, 10 Pogodinskaya str., Moscow, 119121 Russia;
Key words: primers; fluorescein; PCR DOI: 10.18097/BMCRM00194 INTRODUCTION The fluorescently-labeled DNA is widely used in various bioanalytical applications [1-3]. While DNA molecules bearing a single fluorescent tag are mostly employed, the multiple fluorescent labeling can be beneficial since it can enhance the detection sensitivity by increasing a signal-to-noise ratio. Short DNA fragments varying in a degree of labeling can be easily produced during the automated solid-phase chemical synthesis or postsynthetically, whereas the multiple labeling of long DNA fragments is commonly achieved via enzymatic synthesis employing fluorescently labeled nucleoside triphosphates [2]. However, the incorporation of nucleobase-modified nucleotides by DNA polymerases is considerably less effective than that of natural ones, even in the case of primer extension (PEX) on an unmodified DNA template [4]. When the polymerase has to read through the modified template such as in the case of polymerase chain reaction (PCR), the incorporation efficiency can decline dramatically [5]. Moreover, for Taq polymerase (the most widely used DNA polymerase in PCR), the use of modified nucleotides often leads to truncated PCR products [5]. Another recently suggested method for DNA fluorescent labeling relies on base excision trapping, employing deaminated DNA bases to mark label positions, which are excised by base excision repair enzymes generating abasic sites [6]. Yet, this method requires specially designed aminooxy-substituted rotor dyes which are not commonly available. Also, multiple fluorescent tags can be efficiently introduced into DNA postsynthetically by using clickable deoxyuridines incorporated into DNA chains via PEX or PCR ([7] and references therein). However, such postsynthetic DNA functionalization requires a researcher to have particular skills and an access to specific reagents. Interestingly, the compaction of DNA labeled with a single fluorophore can as well increase the overall fluorescent intensity [8], though this approach is rather unpractical since it assumes the use of concentrated polyethylene glycol solutions. As a technically simple alternative, the multiple labeling of long DNA can be carried out by PCR, employing primers with more than a single fluorescent tag. In ideal, the primers have to bear a fluorescent tag at each and every nucleobase where a fluorophore can be potentially attached. In practice, primers with fluorescent tags conjugated to thymine bases are most easily produced as a result of chemical synthesis or postsynthetic modification and routinely available from the commercial companies specializing in oligonucleotide production at the affordable cost. However, it is still unclear whether the “heavily labeled” primers can be employed to successfully produce PCR products bearing multiple fluorescent tags. To answer this question, we tested a set of primers carrying multiple fluorescein tags and varying in a degree of labeling. Fluorescein has been chosen as the most broadly used fluorescent dye utilized presently for DNA labeling. MATERIALS AND METHODS All chemicals were purchased from “Merck” (USA), if not stated otherwise, and were of ACS grade or higher. Solutions used were prepared with Milli-Q quality water (18 MΩ·cm). The primers with 6-carboxyfluorescein moieties (FAM) attached to thymine bases were synthesized by “Syntol” (Russia) and listed in Table 1. The 210 base pair (bp) long DNA fragments employed as DNA template (5’-gctggccagtttgctaccttccagtgcagtgccatcggcaggaccgtggcaggagacaggctctggttacagggcattgatgtgcgagatgctcctctgaaggaaa tcaaggtgaccagctcccgacgcttcattgcttcatttaatgttgtgaataccaccaaacgagatgctggaaagtaccgctgcatgattcgcactgaaggaggt-3’) were produced by PCR from cDNA of transcripts of the human PTPRM gene and purified as earlier described [9]. Real-time PCR was carried out on a DTprime 5M6 thermal cycler (“DNA-Technology”, Russia), using the PCR reaction mixture 5X qPCRmix-HS SYBR (“Evrogen”, Russia) with Taq polymerase. Amplification started with a polymerase activation step (6 min at 95ºC), followed by 40 cycles of subsequent 45-s incubations at 95ºC, 60ºC (unless indicated otherwise), and 72ºC. Concentration of each primer was 0.4 µM. The template amount was 100 amole per PCR reaction. PCR generated DNA amplicons were subjected to electrophoresis in 8% polyacrylamide gel in TBE-buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 8.3). The electrophoresed fluorescent DNA amplicons were visualized by scanning gels on a Typhoon FLA 9500 Gel Imager (“GE Healthcare”, USA), after their staining with SYBR Green I fluorescent dye (“Thermo Fisher Scientific”, USA). The GeneRuler Ultra Low Range DNA Ladder (“Thermo Fisher Scientific”) was used as DNA size standards. RESULTS AND DISCUSSION As an initial step, we have tested a pair of primers of arbitrary sequence where all thymine bases were modified with FAM (primers F*(1) and R*(1), Table 1) and found out that primers labeled at such degree give no products in PCR, whether they were used as a pair (data not shown) or in a pair with the unmodified primer (Figs. 1 and 2). To further investigate how the presence of multiple fluorescein tags can affect the template amplification in PCR, we employed a set of primers differing in labeling at or near the primer 3′-end (Table 1).
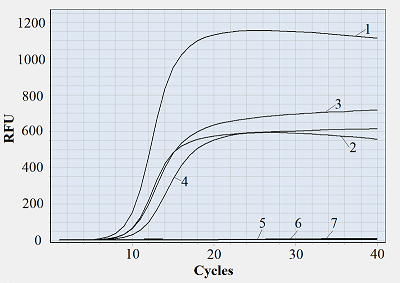
It appears reasonable to vary labeling of primers starting from the primer 3′-end since that part of primer sequence is the most responsible for polymerase binding and primer extension [10]. Figure 1 shows representative amplification curves for different combinations of forward and reverse primers. As seen, if at least one of the primers bears a fluorescein tag attached to a 3′-end nucleotide, no amplification is observed. There was also no amplification when one of the tags was attached to the second nucleotide (from the 3′-end) of a primer while another primer was unlabeled. Only when fluorescein tags were positioned not closer to the 3′-end than at the fourth nucleotide, the amplification occurred (Fig. 1, the F/R*(4) primer pair). Moreover, if both primers are multiply labeled but at positions at least three nucleotides away from the 3′-end, the amplification proceeds (Fig. 1). For all tested pairs of labeled primers where amplification occurred, the overall yield of PCR products (characterized by the level of fluorescence at the upper plateau of amplification curve) was evidently lower compared to that for unlabeled ones. Besides, a shift of Cp values from 9.5-9.7 (for F/R) to 9.8-10.2 (for F*(6)/R and F/R*(4)) and further to 11.0-11.4 (for F*(6)/R*(4)) was found (as illustrated by the shift of amplification curves in Fig. 1)
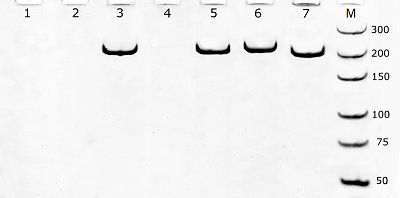
The electrophoretic analysis confirmed that PCR products with size matching the expected (210 bp) were generated during amplification (Fig. 2). It should be mentioned that slight differences in electrophoretic mobility of amplicons, visible in Fig. 2, well agree with the expected higher retardation of heavily modified DNA in polyacrylamide gel electrophoresis [11].
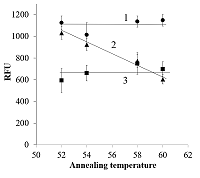
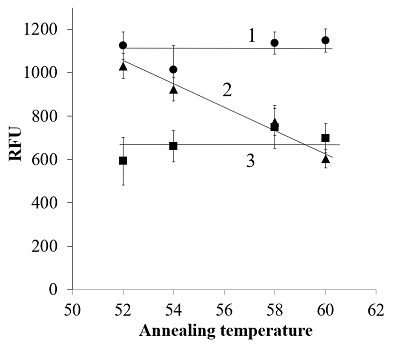
One may assume that a difficulty in polymerase binding at the primer-template junction with a modified 3′-nucleotide can be responsible for a block of DNA amplification. Indeed, the presence of a bulky moiety attached to the 3’-nucleotide of primer can potentially create a steric hindrance for polymerase binding at the primer-template junction thus hindering DNA polymerization. However, when fluorescently labeled nucleotides are incorporated into DNA by PEX [2], the polymerase appears to tolerate the fluorescein tag on the last nucleotide of an extending DNA strand (provided that the polymerase has already bound). Besides, no amplification was also observed when the tag was removed from the primer 3′-nucleotide but retained on the adjacent nucleotide (Figs. 1 and 2, the F*(2)/R primer pair) thus pointing out that the reason behind the observed block of DNA amplification could be more complex. It is known for a long time that mismatches between primers and a template affect the stability of the primer-template duplex and, consequently, the efficiency with which the polymerase extends the primer [12, 13]. Furthermore, mismatches located in the 3′-end region (defined as the last 5 nucleotides at the primer 3′-end) of a primer have significantly larger effects on amplification efficiency than mismatches located closer to the primer 5′-end (e.g., [12]). By the same token, a fluorescein moiety can potentially interfere with proper nucleotide base pairing thus locally destabilizing primer-template duplex. If so, one may expect that a decrease of annealing temperature (Ta) should reduce the observe negative impact of primers’ labeling at or near the primer 3’-end on amplification. Yet, it has been found that the gradual decrease of Ta from 60°C down to 52°C had no effect for primer pairs F*(1)/R, F*(2)/R, and F/R*(1) – no amplification was observed (data not shown). For primer pairs F/R, F*(6)/R, and F/R*(4), no appreciable effect of Ta was found in terms of Cp (data not shown) while the yield of PCR products (estimated as a value of relative fluorescence at the 40-th cycle) demonstrated a clear increase with decreasing Ta for the pair F/R*(4) (approaching that of PCR products for unmodified primers at 52°C) and no dependence within the experimental scatter for pairs F/R and F*(6)/R (Fig. 3). Thus, for the primer R*(4), the obtained result supports the assumption that the presence of a modified nucleotide (which is within the 5 nucleotide long 3’-end region) can destabilize the duplex structure near the primer-template junction, consequently hindering amplification. However, it is still unclear why the PCR yield for the primer pair F*(6)/R at the decreased Ta (52-54°C) is consistently lower than that for the F/ R*(4) pair (Fig. 3).
CONCLUSIONS The positioning of fluorescein tags at or near the 3’-end upon primer multiple labeling can inhibit DNA template amplification (up to a complete stop when tags are placed on the 3’-terminal or adjacent nucleotide). The mechanism, by which the presence of fluorescein tags at or near the primer 3’-end affects the PCR performance, is rather ambiguous and can involve both a steric hindrance from the fluorescein moiety for polymerase binding and destabilization of a primer-template duplex. Nonetheless, if multiple fluorescein tags are attached so that at least three nucleotides from the primer 3’-end are unmodified, the production of DNA fragments bearing multiple fluorescein molecules is possible even if both primers are heavily labeled, though on the expense of yield of amplicons. COMPLIANCE WITH ETHICAL STANDARDS This article does not contain any research involving humans or using animals as objects. FUNDING The study was performed within the framework of the Program for Basic Research in the Russian Federation for a long-term period (2021–2030) (No. 122030100170-5). CONFLICT OF INTEREST The authors declare no conflict of interest. REFERENCES
|