|
Программа предсказания времени удержания пептидов с учётом посттрансляционных модификаций
Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, Ключевые слова: время удержания пептида; изоэлектрическая точка; посттрансляционные модификации; web-сервис DOI: 10.18097/BMCRM00196 ВВЕДЕНИЕ В экспериментах по протеомике время удерживания пептидов на хроматографической колонке (retention time, RT) – важный показатель, который определяется только свойствами самого пептида и особенностями оборудования для жидкостной хроматографии и не зависит от метода фрагментации и способов последующей идентификации [1]. Это даёт возможность использовать данный параметр для оценки достоверности идентификации пептидов [2]. Кроме того, предсказание RT можно использовать при планировании эксперимента для более прицельного отбора пептидов для последующего анализа. Величина RT, наблюдаемая в каждом конкретном эксперименте, зависит от множества факторов даже при работе на одном и том же оборудовании: различий в настройках прибора и характеристик колонки, температурного режима, состава смеси [2]. Однако это препятствие можно обойти, если использовать не непосредственное предсказание величины RT, а предсказание более фундаментальной характеристики пептида, от которой в свою очередь зависит RT. Например, Krokhin и соавторы предложили использовать величину «гидрофобности» (Hydrophobicity Index, HI) и создали программу Sequence-Specific Retention Calculator (SSRCalc) для предсказания данной величины [3], которая использует аддитивную схему с коррекцией. Первоначальная сумма величин, названных коэффициентами удержания, индивидуальных для каждого из аминокислотных остатков в зависимости от его положения в полипептидной цепи, корректируется набором коэффициентов. Эти коэффициенты связаны с размером пептида, зарядом, величиной изоэлектрической точки, наличием более одного остатка пролина рядом, способностью образовывать гидрофобные кластеры или устойчивые спирали. Существуют и другие алгоритмы предсказания RT от простых линейных регрессионных моделей, базирующихся на свойствах аминокислот до сложных биофизических моделей, либо использующие методы глубокого обучения (deep learning). Так, авторы SSRCalc использовали методы глубокого обучения в своей новой программе Chronologer [4]. При этом, Chronologer может предсказывать RT и для пептидов с некоторыми посттрансляционными и химическими модификациями. В данной работе тот факт, вносятся ли эти модификации искусственно или в процессе жизнедеятельности, не существенен, и оба варианта будут обозначены, как ПТМ. Тем не менее, несмотря на видимый выигрыш в точности предсказания, методы глубокого обучения продолжают оставаться «чёрным ящиком», так как нет возможности интерпретировать полученную модель с точки зрения физико-химических свойств объектов. В свою очередь, такая величина как гидрофобность может быть использована сама по себе для предсказания каких-либо других свойств, связанных с пептидами [5], а, например, коэффициенты удержания для отдельных остатков могут быть дескрипторами в QSAR-моделях. Таким образом, представляется полезным добавить в программу SSRCalc возможность предсказания величины HI для пептидов с ПТМ, которой в исходном варианте нет. В качестве базовой была выбрана оригинальная программа SSRCalc версии 3.0, написанная на языке Perl и доступная в рамках Artistic License. Подробности алгоритма программы и используемые коэффициенты опубликованы в работе [3]. Добавочные параметры для остатков с ПТМ могут быть рассчитаны на основании наборов данных масс-спектрометрического анализа. МЕТОДИКА Набор целевых значений для пептидов, идентифицированных при анализе масс-спектрометрических данных Для формирования выборки пептидов для анализа и тестирования были использованы полученные ранее (с использованием программы Peaks Studio X Pro [6]) в работе [7] результаты по идентификации пептидов для 25 наборов исходных (RAW) данных масс-спектрометрических экспериментов, выполненных с применением метода изоэлектрического фокусирования (IEF) и депонированных в БД ProteomeXchange (PXD000065 [8], PXD005410 [9], PXD006291 [10], PXD010006 [11] и PXD017201 [12]). В части из этих экспериментов авторы работ использовали TMT (Tandem Mass Tag) или iTRAQ (Isobaric Tags for Relative and Absolute Quantification) метки (только одна выборка c iTRAQ из двух была использована в работе, причина описана далее), в 6 случаях метки не использовали. В каждом наборе данных были представлены 72 фракции, отобранные при в узком поддиапазоне pH. В отличие от работы [7], из общего массива идентифицированных пептидов отбирали только те, которые удовлетворяли двум условиям: уровень FDR 0.1% и значение ASCORE при наличии модификации не менее 500. В работе [7] ограничение для идентификации прекурсорного иона было установлено в 5 ppm, точность идентификации фрагментов – 0.01 Da. Все модификации, включая алкилирование остатков цистеина и наличие меток TMT или iTRAQ, считали вариабельными. Это позволило ожидать, что часть непрореагировавших с советующими реагентами пептидов также будут зарегистрированы. Кроме того, были наложены дополнительные ограничения. Пептид не должен был быть идентифицирован менее чем в 2-х соседних фракциях (отличаются по pH), но и не более чем в числе фракций, соответствующих диапазону pH шириной 0.3 для экспериментов с широким диапазоном pH и 0.1 с узким. Кроме того, был использован дополнительный набор данных из работы [13], в которой каждая фракция была получена из фрагмента 2D геля. Это позволило получить данные для описание дополнительной модификации – образование пропионамида, а также позволило добрать данные по пептидам с оксипролином. Фильтрацию по диапазону pH в последнем случае не использовали. Описание каждого из наборов данных представлены в таблице 1.
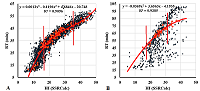
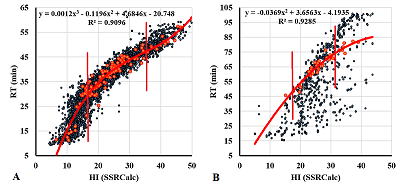
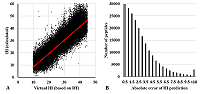
Как и можно было ожидать, число непрореагировавших пептидов в экспериментах с использованием меток очень мало. В то же время число немодифицированных пептидов в конкретной фракции имеет определяющее значение для второго этапа подготовки данных – пересчёта значений RT в величину гипотетической гидрофобности (virtual HI) на основе корреляции наблюдаемых величин RT и рассчитанных программой SSRCalc для каждой отдельной фракции (рис. 1). Для построения корреляционного уравнения было установлено ограничение: использовали только данные для немодифицированных пептидов в пределах одной фракции, не менее 7 уникальных последовательностей с разницей между минимальным и максимальным значением RT не менее 15 минут. Для создания корреляционных уравнений с модификацией TMT отбирали только непрореагировавшие пептиды без модификаций, имеющие аналоги с ТМТ в той же фракции, исключая этим часть возможных ошибок идентификации. Учитывая, что при сравнении наблюдаемых величин RT и значений HI только в части диапазона значений RT наблюдалась линейная зависимость (рис. 1), то наблюдения, выходящие за пределы этого диапазона, в работе не рассматривали.
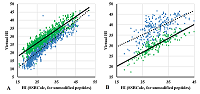
После получения уравнений корреляции для каждой фракции в отдельности по данным наблюдаемого значения RT было рассчитано значение virtual HI. В выборку для подбора коэффициентов удержания отбирали пептиды, имеющие только одну требуемую модификацию в соответствующей позиции (N-концевой остаток, 2-й остаток, «средний» – от 3-го до 2-го с C-конца, предпоследний и C-концевой остаток). Для условно «длинных (от 10 остатков) и условно «коротких» (от 5 до 10 остатков) пептидов выборки формировались отдельно. Учитывали только наборы уникальных пептидов от 8 штук. Подбор коэффициентов удержания проводили в диапазоне от -10 до +20 с шагом в 0.05. Наиболее подходящее значение выбирали по минимальной сумме квадратов отклонений вновь рассчитанных значений Hi от значений virtual HI по всей выборке. В работе также были ограничены варианты модификаций для которых отбирали данные (в скобках указана моноизотопная дельта масс): ТМТ (+229.16) и iTRAQ (+304.21) метки, а также ацетилирование (+42.01), формилирование (+27.99) и метилирование (+14.02) N-концевого остатка и/или бокового радикала лизина; карбамидометилирование (+57.02) остатков цистеина, аспарагиновой и глутаминовой кислот; окисление (+15.99) и двойное окисление (+31.99) остатков метионина и пролина, фосфорилирование (+79.97) остатков серина, треонина и тирозина; С-концевое амидирование (-0.98) остатков лизина и аргинина; образование пропионамида (+71.04) с остатком цистеина. Нет нужды отдельно рассматривать в работе дезамидирование (+0.98) остатков глутамина и аспарагина, так как по сути это иммитирует аминокислотную замену на остаток аспарагиновой и глутаминовой кислот, параметры для которых в программе SSRCalc есть. К сожалению, некоторые из востребованных модификаций, например, фосфорилирование остатка тирозина, при заданных ограничениях не были представлены в выборке в достаточном количестве (8 и более аминокислотных остатков). В таком случае подбор проводили по всему массиву данных, включая пептиды с двумя и более модификациями, например, наличие TMT метки и фосфорилирование остатка тирозина в одном пептиде. Следует отметить, что в случае выборки с iTRAQ меткой, пары пептидов с меткой и без не были обнаружены. Вероятно, процент непрореагировавших пептидов, в отличие от процедуры внесения TMT метки или обработкой йодацетамидом (+57.02), ничтожно мал. Однако в данном случае при вычислении virtual HI можно опереться на загрязнение лабораторного оборудования, т.е. использовать пептиды, не обработанные в пробе, но уверенно определяемые при масс-спектрометрии (рис. 2). Только для одной из двух выборок с iTRAQ меткой из работы [7] таких данных было достаточно для подбора уравнения пересчёта RT в virtual HI. В данном случае все фракции рассматривали как единое целое, так как данных по примесям было не так много. Однако если сравнивать между собой значения RT для одинаковых пептидов из разных фракций, то для данной выборки отклонения в среднем не превышали одной минуты.
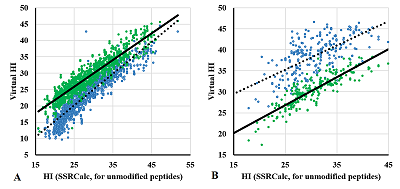
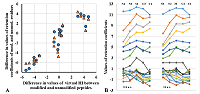
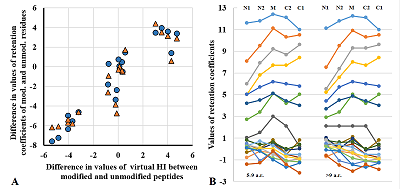
Программа Retention Time Predictor (RTP) - модификация программы SSRCalc для работы с учётом ПТМ Оригинальный код программы SSRCalc v3.0 на языке Perl был транслирован на язык Python (версия 3.6.8). Воспроизводимость результатов для немодифицированных пептидов была проверена на выборке из более чем 215 тысяч уникальных пептидных последовательностей, собранных из всех 25 выборок, рассматриваемых в работе. В случае округления результатов до второго знака после запятой при записи в файл максимальное отклонение в 0.01 единицу HI наблюдали у 239 пептидов, что составило 0,11% от общей выборки. Остальные значения были идентичны. Наличие небольшой ошибки округления вероятнее всего является следствием различий в округлении дробных чисел в языках Perl и Python. В соответствии с алгоритмом оригинальной программы SSRCalc, вычисление параметра HI происходит в два этапа. Первоначально суммируются табличные коэффициенты удержания для каждого из аминокислотных остатков пептида в зависимости от их расположения в пептиде. Различаются позиции N-концевая (N1), вторая с N-конца (N2), любая в середине цепи (M), предпоследняя с С-конца (С2), С-концевая (С1). На втором этапе последовательно рассчитывается ряд поправочных коэффициентов, в зависимости от аминокислотного состава и свойств пептида, его длины и т.д. Итоговая величина является гидрофобностью пептида. Разработчики SSRCalc [3] описали 2 шкалы коэффициентов удержания для колонок с размером пор 10 нм и 30 нм, в настоящей работе использовали последнюю. Способ учёта модифицированных остатков при вычислении поправочных коэффициентов определяли из общих соображений, как могут измениться свойства остатков при внесении данной модификаций. Например, остаток метионина при расчёте поправки «кластерность» определён как сильно гидрофобный, следовательно, его окисленная форма не учитывается при расчёте данной поправки. В тоже время остаток оксипролина своей геометрии не меняет и при расчёте поправки на наличие полипролинового фрагмента продолжает учитываться. При расчёте поправок, связанных с наличием заряда на остатке, учитывали изменения в числе протонируемых или диссоциируемых групп для данной модификации. Так как табличных данных по значениям pKa для расчёта величины pI модифицированных пептидов в программе SSRCalc нет, то для её расчёта программа RTP использует шкалу программы pIPredict версии 3 [7] (вариант без учёта соседних остатков). При тестировании влияния замены алгоритма расчёта pI на той же выборке из более чем 215 тысяч немодифицированных пептидов среднее отклонение составило 0.04 единицы HI, максимальное – 0.82, при этом среднее отклонение величины pI было 0.32 единицы pH, максимальное – 4.25. Между программами имеются также различия в описании остатков, связанных c модификациями N-концевой аминогруппы. Они трактуются как добавочный остаток, для которого имеются собственные табличные данные. Например, пептид A(+229.16)GGSTR в программе преобразуется в форму [+229.16]AGGSTR, где [+229.16] – самостоятельный остаток с массой 229.16 и собственными табличными значениями коэффициентов удержания, а остаток аланина становится вторым. Это позволяет сократить размеры шкалы. В свою очередь пептид K(+229.16)(+229.16)GGSTR преобразуется к виду [+229.16]K(+229.16)GGSTR, где K(+229.16) – остаток с массой 357.26 (с модифицированным боковым радикалом), также имеющий собственные табличные значения коэффициентов удержания. Если программа встречает пептид K(+229.16)GGSTR, то трактует его как пептид с модификацией N-концевой аминогруппы, а не бокового радикала. Строго говоря, последнее правило может не соответствовать действительности, однако, способа различить на каком из атомов азота первого остатка находятся модификации [+304.21], [+229.16] или [+27.99] нет. Для модификации [+42.01] и [+14.02], если пептид не соответствует N-концевому пептиду белка, можно предположить, что модифицирован боковой радикал, но всё равно применятся данной правило. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ В таблице 2 указано количество отобранных наблюдений в подвыборках для пептидов с различными модификациями и по каждой из возможных позиций. Первая величина – количество наблюдений, когда в каждом пептиде модифицирован только 1 остаток, величина в скобках – количество пептидов, содержащих данную ПТМ. В случае одиночной ПТМ хорошо видно согласованное изменение гидрофобности пептидов (рис. 2). При этом направление изменений хорошо согласуется с ожидаемым результатом, что и должно быть для аддитивной схемы расчёта при изменении единственного параметра. В общем случае можно просто вычислить среднюю величину изменений (или лучше величину смещения линии тренда) и добавить её к значению коэффициента удержания немодифицированного остатка; результат будет близким к величине, подобранной перебором на всех пептидах с данной ПТМ (рис. 3А). Расчёт тех же величин на выборке пептидов, включающих более одной ПТМ, даёт близкие величины. Таким образом, можно ожидать, что и для случая, когда нет достаточного набора данных с одиночными ПТМ, результат будет адекватный.
Подобранные значения коэффициентов удержания приведены в таблице 3. К сожалению, некоторые из модификаций не имеют достаточного числа наблюдений по отдельным позициям ни в одном из вариантов (одиночные или множественные модификации). Но если проанализировать таблицу значений для немодифицированных остатков (рис. 3B), то в среднем минимальное и максимальное значение коэффициентов удержания отличаются на 1.7 единиц, так что в некотором приближении можно взять среднюю величину по имеющимся значениям и заполнить таблицу полностью. На анализируемых в этой работе данных ошибка в 1 единицу величины HI приводила к ошибке при определении величины RT в 1-3 минуты. При этом величина RT по сути дискретная, так как определяется с некоторым шагом, а минимальное среднее отклонение от линии тренда при сравнении величин HI и RT для немодифицированных пептидов для отдельных фракций составило около 3 минут.
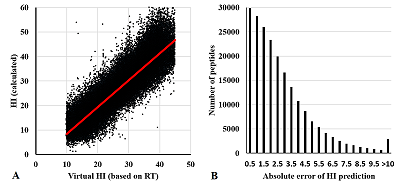
Рисунок 4 демонстрирует зависимость величины virtual HI, вычисленной из RT, и величины HI, рассчитанной с учётом имеющихся ПТМ, на всём массиве пептидов с ПТМ (около 200 тысяч), использованных в работе. Для 90% наблюдений абсолютная ошибка не превышает 5. Следует напомнить, что расчёт величины virtual HI с использованием корреляционных уравнений может вносить определённую ошибку. Часть значений соответствуют пептидам с возможной ошибочной идентификацией (с учётом выбранного уровня FDR их не меньше 1%), что соответствует количеству тех пептидов, для которых абсолютная ошибка больше 10. Это можно использовать для фильтрации пептидов с ложной идентификацией.
Программная реализация, доступность кода и ограничения при работе программы. Для расчёта величины HI с использованием полученной шкалы значений коэффициентов удержания можно использовать программу RTP, свободно доступную для пользователей. В функции программы RTP входит также предсказание величины RT по коэффициентам уравнения корреляции в виде многочлена до пятой степени (вводятся пользователем). В процессе выполнения программы для немодифицированных пептидов возможен выбор метода предсказания pI (с помощью шкалы pIPredict 3 или оригинальной шкалы SSRCalс). Так как для немодифицированных пептидов изменения в шкалу значений коэффициентов удержания не вносили, то для таких пептидов программа повторяет вычисления SSRCalс версии 3. Программа RTP доступна через веб-интерфейс по адресу http://lpcit.ibmc.msk.ru/RTP, а также в виде исполняемого файла для операционной системы MS Windows 10 (по ссылке на той же странице). Код программы может быть предоставлен по запросу. Программа работает с входными файлами в формате FASTA или TXT, в котором пептиды располагаются построчно без заголовков. Описание ПТМ соответствует выходным файлам программы Peaks Studio X Pro. Для веб-интерфейса установлено ограничение при загрузке в 10000 пептидов, размер файла не более 900 кбайт. В случае превышения лимита, данные будут загружены не полностью, и обработаны будут только первые 10000 пептидов. Результаты представлены в виде таблицы, содержащей последовательность пептидов, предсказанные значения HI, pI и RT (последнее при условии ввода коэффициентов уравнения). Результаты также доступны в виде текстового файла с разделителем «табуляция». Формируется гистограмма распределения значений предсказанной величины Hi или RT. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Данная работа не содержит каких-либо исследований с использованием людей и животных в качестве объектов исследования. ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021 - 2030 годы) (№ 122030100170-5). КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|