|
Усовершенствование экзонового метода для ускоренного получения кДНК гена реналазы крысы
Научно-исследовательский институт Биомедицинской химии им. В.Н. Ореховича, Ключевые слова: ген реналазы крысы; экзон; объединение экзонов; ПЦР; экспрессия гена DOI: 10.18097/BMCRM00201 ВВЕДЕНИЕ
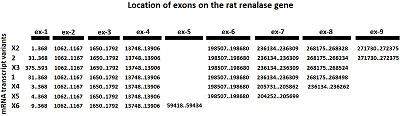
Использование стандартной процедуры получения ДНК ампликонов любых участков геномной ДНК эукариот и последующее объединение таких участков ДНК позволяет конструировать любые генетические конструкции с известной нуклеотидной последовательностью [1-6]. В данной работе мы усовершенствовали разработанный нами ранее метод объединения отдельных кодирующих последовательностей (экзонов) геномной ДНК при получении кДНК гена, которое проводили в несколько этапов, попарно наращивая кодирующую последовательность до получения полноразмерной кДНК [5, 6]. При получении кДНК в данной работе был апробирован метод объединения сразу нескольких (четырех) экзонов. Ген крысиной реналазы (Rattus norvegicus, код доступа GenBank NC_051336.1), локализован на хромосоме 1 и содержит 272375 пар оснований (п.о.). В результате вычислительного анализа с использованием метода Gnomon (gene prediction method Gnomon) в гене реналазы крысы было определено девять экзонов, которые включают семь транскрипционных вариантов мРНК: 1, 2, X2, X3, X4, X5, X6. Транскрипционные варианты мРНК реналазы содержат экзоны, в которых (в первом, седьмом и восьмом экзонах) транскрипционные области по величине незначительно варьируют. Транскрипционные варианты мРНК Х2 и 2 включают восемь экзонов, транскрипционные варианты мРНК Х3, 1 и Х4 включают семь экзонов, транскрипционный вариант мРНК Х5 содержит шесть экзонов, а транскрипционный варианты мРНК Х6 – пять (рис.1). Для синтеза кДНК гена реналазы крысы нами был выбран транскрипционный вариант-2 мРНК, содержащий 8 экзонов. МЕТОДИКА В работе были использованы следующие реагенты: олигонуклеотиды для ПЦР, (табл.1) синтезированные компанией «Евроген» (Россия), ДНК полимеразу Tersus Plus PCR kit ™ («Евроген»), маркеры молекулярной массы, синтезированные в нашей лаборатории, эндонуклеазы рестрикции BamHI, XhoI и Ni-Sepharose («Cytiva», США). Система для очистки фрагментов ДНК Wizard® SVGel и PCRClean-Up и TA-вектор (pGEM®-T-easy vector) получены от «Promega» (США). Плазмидный экспрессионный вектор pET-28a(f+) и клетки E. coli штамм Rosetta (DE3) были получены от «Novagen» (Великобритания). Другие химические реактивы были приобретены у «Sigma-Aldrich» (США).
Геномную ДНК выделяли из замороженных образцов крови крыс линии WKY. Процедура выделения геномной ДНК из лимфоцитов крови крысы не отличалась от процедуры выделения геномной ДНК из лимфоцитов человека [5, 6]. Амплификацию экзонов проводили в 20 мкл реакционной смеси содержащей: 6,7 мМ Tris HCl pH 8,8; 1,66 мМ (NH4)2SO4; 0,02% Tween 20; 1 мМ MgCl2; 0,2 мМ dNTP; 1% глицерин, по 4 мкМ каждого праймера, 50 – 100 нг геномной ДНК и 0,5 ед. Tersus ДНК полимеразу. Условия ПЦР: денатурация – 95ºС в течении 5 мин. – 1 цикл; элонгация – 92ºС – 20 с, 55ºС – 15 с, 72ºС – от 30 с до 2-3 мин; количество циклов от 30 до 35; заключительный цикл 72ºС – 3 мин. Время элонгации с ДНК полимеразой PfuUltra зависело от длины синтезируемого фрагмента. Синтез одиночных экзонов во время элонгации проводили 30-40 с, а полноразмерной кДНК (при объединении всех экзонов) – 3-4 мин. При реамплификации матрицы ДНК использовали те же условия, матрицей служил 1 мкл ДНК ампликон от предыдущего этапа амплификации. Электрофорез в 2% агарозном геле и электрофорез в 12% полиакриламидном геле (ПААГ), клонирование кДНК гена реналазы, трансформация клеток E. coli ДНК вектором, экспрессия клонированного гена описаны в наших предыдущих работах [5, 6]. Секвенирование ДНК проведено компанией «Евроген». Выделение рекомбинантного белка, содержащего гексагистидиновую метку, осуществляли на Ni-Sepharose в полном соответствии с ранее приведенным протоколом [7]. РЕЗУЛЬТАТЫ Транскрипционный вариант-2 мРНК реналазы крысы, содержащий 1802 нуклеотидов, включает восемь основных экзонов (код доступа- GenBank NM_001014167) (рис. 1) и кодирует белок, содержащий 315 аминокислотных остатков (а.о). Для получения полной кДНК последовательности реналазы методом ПЦР на матрице геномной ДНК крысы было синтезировано восемь отдельных экзонов с использованием праймеров, приведенных в таблице 1.
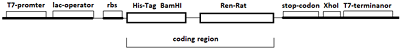
Выбор праймеров При клонировании гена крысиной реналазы с применением экзонового метода, на матрице геномной ДНК крысы методом ПЦР вначале получали отдельные кодирующие области семи экзонов (ex-1, ex-2, ex-3, ex-4, ex-6, ex-7 и ex-8+ex-9) (рис. 2). Короткая кодирующая область ex-9 транскрипционного варианта-2 мРНК реналазы составляла 12 нуклеотидов и поэтому ее получали при синтезе ex-8 в составе обратного праймера, содержащего ex-9 (ex-8+ex-9). Исходя из этого, для синтеза 7 кодирующих областей гена реналазы (экзонов) методом ПЦР было синтезировано 14 праймеров (семь прямых и семь обратных) (табл. 1). При синтезе н.п., кодирующей области первого экзона (ex-1), прямой праймер (1RR-for– forward) содержал начальную фланкирующую область с сайтом рестрикции BamHI, необходимую для встраивания в вектор pET-28a(+), и концевую область, состоящую из 20 нуклеотидов ex-1. Обратный праймер (1RR-rev – reverse) содержал 20 нуклеотидов концевой части первого экзона. При синтезе 6 оставшихся экзонов, прямые праймеры (2RRe-for–8RRe-for) также содержали две нуклеотидные области. Начальная нуклеотидная область включала 20±3 нуклеотидов конца предыдущего экзона; она была областью перекрывания между соседними экзономи и служила для их объединения. Концевая нуклеотидная область состояла из 20±3 нуклеотидов начала амплифицируемого экзона. Обратные праймеры (1RRe-rev–7RR-rev), содержали 20±3 нуклеотидов комплементарной концевой нуклеотидной последовательности (н.п.) амплифицируемого экзона. Длину праймера подбирали в зависимости от A-T/G-C состава н.п. кодируемой области экзонов. Обратный праймер последнего экзона (8-9RR-rev) содержал добавочную фланкирующую н.п. (15 нуклеотидов) 8 экзона, нуклеотидную последовательности 9-го экзона (9 нуклеотидов), терминирующий кодон (TGA) и сайт рестрикции XhoI (CTCGAG), необходимый для встраивания в плазмидный вектор pET-28a(+) (табл. 1).
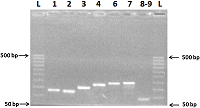
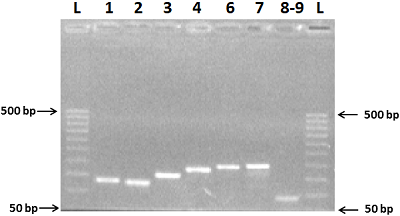
Синтез экзонов Синтез кодирующей н.п. (кДНК) гена реналазы крысы проводили методом ПЦР на матрице геномной ДНК, полученной из лимфоцитов цельной крови крысы. Вначале на матрице ДНК методом ПЦР получали ДНК ампликоны от восьми экзонов (ex-1, ex-2, ex-3, ex-4, ex-6, ex-7 и ex-8+ex-9). При синтезе экзонов использовали праймеры: ex-1 - 1RR-for и 1RR-rev; ex-2- 2RR-for и 2RR-rev; ex-3 - 3RR-for и 3RR-rev; ex-4- 4RR-for и 4RR-rev; ex-6 - 6RR-for и 6RR-rev; ex-7 --for и 7RR-rev; ex-8 совместно с ex-9 - 8RR-for и 8-9RR-rev. Амплификация одиночных экзонов на матрице геномной ДНК давала хороший выход целевого продукта (Рис. 3). В результате амплификации тотальной матрицы ДНК одиночных экзонов в раздельных ПЦР с использованием соответствующих праймеров были получены ДНК ампликоны от восьми экзонов с расчетным размером в парах оснований (п.о.): ex-1 - 135 п.о., ex-2–128 п.о., ex-3 – 159 п.о., ex-4 – 182 п.о., ex-6 –199 п.о., ex7 –196 п.о., ex-8+ex-9 –92 п.о. (рис. 3).
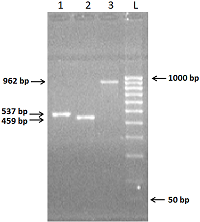
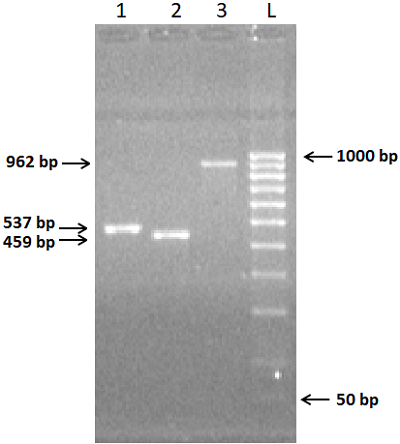
Сборка экзонов в полноразмерную кДНК Последующее объединение соседних экзонов проводили не попарно, как делали ранее [5, 6], а объединяли сразу по четыре экзона. Такое объединение возможно, поскольку 5'-конец прямых праймеров, используемых для амплификации каждого экзона, содержал последовательность 20 ± 3 нуклеотидов, комплементарную началу предыдущего экзона. При синтезе полноразмерной кДНК гена реналазы крысы мы не стали проводить попарное объединение соседних экзонов (1-й со 2-м, 3-й с 4-м, 6-й с 7-м), как делали ранее при синтезе кДНК генов реналазы 1 и 2 человека [5, 6]. Это позволило сократить процедуру сборки кДНК за счет одностадийного этапа объединения четырех экзонов: ex-1, ex-2, ex-3 и ex-4 объединяли с использованием праймеров 1RR-for и 4RR-rev, а экзоны ex-6, ex-7 и ex-8 + ex-9 – с использованием праймеров 6RR-for и 8-9RR-rev (см. таблицу 1). Используя праймеры 1RR-for и 8-9RR-rev (Табл. 1), осуществляли объединение ДНК ампликонов, содержащих с 1 по 4 экзон с ДНК ампликонами, содержащими с 6 по 9 экзон. В результате была получена полная кодирующая нуклеотидная последовательность кДНК, соответствующая транскрипционному варианту-2 мРНК реналазы крысы (рис. 4).
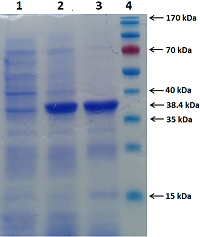
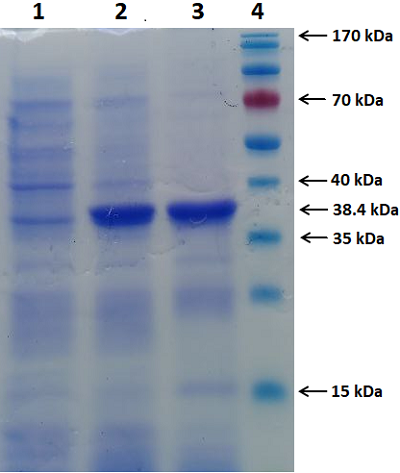
Клонирование и экспрессия Полученную кДНК последовательность встраивали в прокариотический вектор pET-28a(+) по сайтам рестрикции BamHI/XhoI. В результате такого встраивания к N-концу целевого рекомбинантного белка реналазы добавляется гексагистидиновая последовательность (6His-Tag) и аминокислотная последовательность от вектора pET-28a(+) (в общей сложности к целевому белку добавляется N-концевой пептид, состоящий из 34 а.о.). На С-конце нуклеотидной последовательности был исключен 6His-Tag остаток посредством ввода терминирующего кодона. Таким образом, встроенная в прокариотический вектор pET-28a(+) генетическая конструкция кодирует рекомбинантный белок реналазы крысы в 349 а.о. с расчетной молекулярной массой 38.4 кДа (рис. 2). Для клонирования полученную кДНК гена реналазы крысы размером 962 п.о. очищали электрофоретически на агарозном геле, затем проводили экстракцию и очистку системой очистки фрагментов ДНК (Wizard), следуя протоколу производителя. Очищенный фрагмент ДНК встраивали вначале в плазмидный вектор pGEM-T по сайтам рестрикции BamHI/XhoI. В результате клонирования плазмидного вектора в клети E. coli было отобраны клоны, содержащие рекомбинантную ДНК в векторе pGEM-T. В результате рестриктного анализа рекомбинантного вектора pGEM-T были отобраны 3 клона со вставкой ДНК в области 962 п.о. Далее был проведен сиквенс-анализ отобранных клонов. Секвенирование н. п. рекомбинантных векторов было выполнено компанией «Евроген». Секвенированную последовательность клонированной ДНК анализировали методом BLAST анализа («National Center for Biotechnology Information», США). Он показал полное соответствие полученной н.п. гену реналазы крысы, приведенному в базе данных GenBank (код доступа GenBank NM_001014167). Во всех клонах мы не обнаружили точечных мутаций в клонированной кДНК, которые нельзя было исключить априори на этапе амплификации. Такие рода мутации обычно связаны с недостаточно высокой репликативной точностью работы Taq ДНК-полимеразы. Мы использовали высокоточную Tersus Plus ДНК полимеразу («Евроген»). Tersus полимераза — специально разработанная смесь термостабильных ДНК полимераз для высокоточной и специфичной амплификации фрагментов ДНК. Tersus полимераза обладает высокой точностью синтеза в 5’ → 3’ ДНК-полимеразной активностью (до 10000 п.о.). Ее отличают высокая специфичность амплификации, наличие корректирующей 3’ → 5’ экзонуклеазной активности и отсутствие 5’ → 3’экзонуклеазной активности [8]. Клонированная в вектор pGEM-T кДНК последовательность гена реналазы крысы была вырезана и переклонирована в вектор pET-28a(+) по сайтам рестрикции BamHI/XhoI. В результате переклонирования кДНК последовательности в вектор pET-28a(+) был получен вектор с геном реналазы крысы (кодирующая область мРНК вариант-2), который был обозначен как pET-RenRatV2. Экспрессия гена реналазы крысы в клетках E.coli Экспрессию вектора pET-RenRatV2 осуществляли в клетках E.coli Rosetta (DE3). В результате экспрессии был получен рекомбинантный белок реналазы крысы с расчетной молекулярной массой 38.4 кДа, содержащий на N-конце гексагистидиновый пептид (6His-Tag) и в общей сложности включающий 349 а.о (рис. 5). Очистку экспрессируемого рекомбинантного белка проводили Ni-Sepharose в условиях, описанных нами ранее [7].
ЗАКЛЮЧЕНИЕ При получении кДНК гена реналазы крысы был использован разработанный нами ранее метод экзонового клонирования кДНК эукариотических генов, который позволил упростить получение полноразмерной кДНК. Процедура получения полноразмерной кДНК была сокращена за счет того, что мы использовали объединение сразу нескольких (четырех) соседних экзонов. Такое объединение облегчает получение кДНК эукариотических генов и позволяет получать полноразмерную последовательность кДНК в три этапа: 1) – получение отдельных экзонов; 2) – объединение сразу четырех соседних экзонов; 3) – получение полноразмерной кДНК последовательности. Ранее количество этапов прямо зависело от числа амплифицируемых экзонов [5, 6]. Корректность такого подхода подтверждена путем секвенирования полученной кДНК. Она показала полное (100%) совпадение с н.п., приведенной в базе данных GenBank (код доступа- GenBank NM_001014167). Отсутствие точечных мутаций в клонированной кДНК связано с использованием высокоточной и специфичной для амплификации Tersus Plus ДНК полимераз. Это важный этап при получении ДНК ампликонов, который требует использования высокоточной ДНК полимеразы. Ранее мы использовали ДНК полимеразу PfuTurbo («Евроген»), которая обладала той же точностью, что и ДНК полимеразы Pfu, но более высокой продуктивностью. Однако данная ДНК полимераза вносила точечные мутации, что затрудняло получение кДНК последовательности, соответствующему клонируемому гену. Наши результаты свидетельствуют о том, что при помощи изложенного в данной работе подхода можно ускорить получение кДНК эукариотических генов, содержащих любое количество экзонов. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Данная работа не связана с исследованиями с использованием людей и животных в качестве объектов. ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021–2030 гг.) (№ 122030100170-5). КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|