|
Выравнивание и нормирование данных масс-спектрометрических экспериментов с использованием индекса гидрофобности Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121, Москва, ул. Погодинская, 10; *эл. почта: an.voronina@list.ru Ключевые слова: время удержания; масс-спектрометрия; выравнивание данных DOI: 10.18097/BMCRM00245 ВВЕДЕНИЕ В настоящее время жидкостная хроматография (ЖК), сопряженная с масс-спектрометрией (ЖХ-МС) или тандемной масс-спектрометрией (ЖХ-МС/MC), стала важным инструментом протеомного и метаболомного анализа сложных образцов [1]. Однако существенной проблемой при сравнении результатов нескольких измерений ЖХ-МС, как в технических повторах, проводимых для повышения надежности идентификации, так и в случае анализа нескольких биологических образцов, является вариабельность времени удерживания (RT) в разных экспериментах. Данные ЖХ подвержены влиянию большого числа внешних факторов, в том числе таких, как качество пробоподготовки, незначительных отклонений в настройках оборудования, колебание давления, изменение температуры колонки или подвижной фазы, а иногда и различия в анализируемых пробах. Вариабельность RT на хроматографической колонке встречается во всех наборах данных, поэтому для корректного сравнения образцов часто необходимо скорректировать искажения по RT. Это также важно для обработки данных для количественной безметочной протеомики (ЖХ-МС/MC), когда требуется нормализация данных по интенсивности сигнала [2, 3]. Существует несколько подходов к решению данной проблемы. Первый включает сравнение карт ЖХ-МС на уровне необработанных данных [4] с помощью методов многостороннего анализа данных. Несмотря на то, что эта группа алгоритмов позволяет выявлять дифференциально экспрессированные белки, к недостаткам данного подхода следует отнести большое время работы, невозможность выравнивания для сильно отличающихся биологических проб [1] или разных фракций при многомерном ЖХ-МС разделении и тот факт, что алгоритмы обычно описываются для парного выравнивания. Другой подход – выравнивание карт признаков в ЖХ-МС [1]. Принципиально это очень близкий подход, однако он предусматривает использование не всей совокупности данных, а карты признаков, сгруппированных по заданным параметрам. При этом могут корректироваться как линейные, так и нелинейные искажения величины RT по всем картам признаков. Этот подход широко используется, например, в программах msInspect, MZmine , OpenMS и XCMS и др. [1]. Несмотря на свою быстроту, он требует предварительной обработки данных, а используемые для этого алгоритмы также могут вносить собственные ошибки. Как и предыдущий подход, анализ карт признаков использует данные только ЖХ-МС и так же сильно зависит от сходства между наборами данных ЖХ-МС. Существует и альтернативный метод, объединяющий информацию, полученную с помощью ЖХ-МС, с информацией, полученной с помощью МС/МС [5]. В этом варианте МС/МС-спектры с уверенно идентифицированными пептидными последовательностями представляют собой эталон, относительно которого данные ЖХ-МС(/МС) могут быть выравнены. Считается, что данный подход даёт недостаточно точное выравнивание, и иногда его используют как первый этап (предварительное выравнивание), дополняя затем процедурами, относящимися к первым двум вариантам [6-8]. Основное преимущество данного варианта – возможность выравнивать наборы данных, которые имеют большие различия, например, отдельные фракции после многомерного разделения [5]. Также следует отметить, что точность выравниванияв в этом варианте достаточна для решения широкого спектра задач, например, для контроля ложных идентификаций или сравнения количества конкретного пептида в различных пробах. Последнее также требует нормализации данных в различных пробах по интенсивности сигнала, первым этапом для чего также необходимо выравнивание по RT. Описанные в данной статье алгоритм и программа используют именно этот вариант выравнивания. МЕТОДИКА В работе использованы 2 набора данных ЖХ-МС/МС. Первый взят из работы [9], в которой моделировали ишемию головного мозга с использованием окклюзии средней мозговой артерии. Исходные данные работы [9] доступны в базе данных ProteomeXchange [10] (accession code PXD032141). Использовали только набор данных для молодых самцов мышей с диабетом (выборка 1: всего 24 пробы, 8 из зоны инфаркта мозга, 8 из другого полушария мозга и 8 контрольных, по 4 из разных полушарий мозга). Второй – данные, полученные в работе [11] (accession code PXD000065), в которой для анализа белков клеточной линии A431 был использован метод фракционирования пептидов с использованием изоэлектрического фокусирования (HiRIEF). Использовали часть данных для немодифицированных пептидов, полученных в диапазоне pH от 3 до 10 (выборка 2: 72 пробы с шагом в ~0.1 значения pH). В обоих случаях идентификация пептидов была проведена заново средствами программы PEAKS Studio [12] (погрешность идентификации для первичных ионов 10 ppm и 0.02 Da для вторичных ионов, уровень False Discovery Rate (FDR) - 1%). Каждую пробу анализировали независимо. В качестве входных файлов для последующего анализа использовали файлы экспорта PEAKS Studio “peptide features” (PF). В качестве набора «эталонных» пептидов использовали часть обучающей выборки программы Chronologer [13], в свою очередь взятую авторами из работы [14]. Из данного набора были удалены все пептиды с модификациями и пептиды короче 9 аминокислотных остатков и больше 50. В окончательный набор вошло 364232 пептида с диапазоном значений величины индекса гидрофобности HI (Hydrophobicity Index) от -0.07 до 31 (в шкале «Prosit RT» [15]: от -36.5 до 169.5). В работе использовали величину HI, так как именно её предсказывает программа Chronologer. В случае, если в «эталонном» наборе недостаточно пептидов, совпадающих с исследуемым набором, то можно сформировать новый набор пептидов, относительно которого будет проводиться выравнивание, с использованием предсказанных программой Chronologer величин HI. Также можно использовать и нашу программу RTP [16], предсказывающую ту же величину, но в отличие от Chronologer, использующую простую аддитивную схему расчёта, а не нейронные сети. При аддитивной схеме расчёта средняя ошибка предсказания больше, чем при использовании нейронных сетей, но вероятность случайного выброса очень низкая. При любом варианте предсказания, чем больше пептидов в эталонном наборе, тем меньше влияние ошибки предсказания на конечный результат. При работе с данными, полученными на млекопитающих, должно хватать и имеющегося набора с экспериментальными данными (табл. 1).
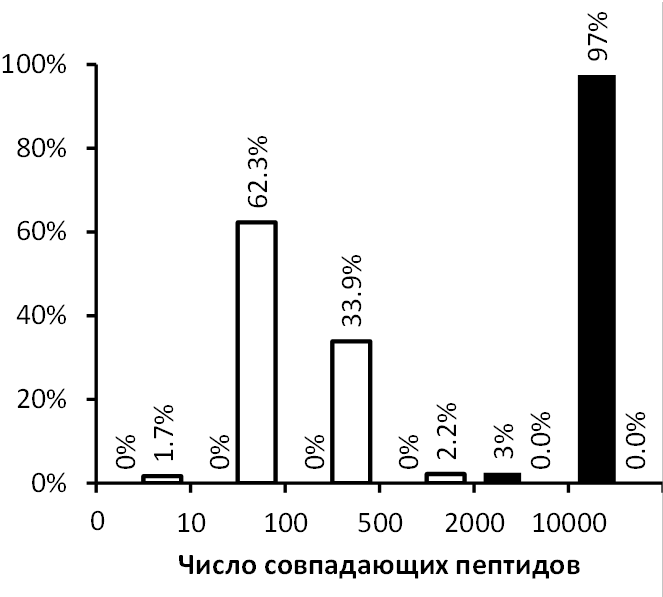
Алгоритм вычислений, описанный ниже, имплементирован в программу, написанную на язык Python (версия 3.6.8) и свободно доступную по адресу http://lpcit.ibmc.msk.ru/AlignRT. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Выравнивание данных относительно «эталонного» набора. Строго говоря, для выборки 1 (как и в других подобных случаях, когда данные получены на одной конкретной ткани, пусть и для различных условий) в качестве эталона можно использовать данные любой из собственных проб выборки, так как любые 2 пары проб включают в себя не менее нескольких тысяч совпадающих пептидов (рис. 1), относительно равномерно распределённых по значениям RT. Как правило, так и делают. В таком случае использование универсального стандарта полезно для последующего сравнение с данными, полученными на образцах других тканей. Причём сравнивать можно сразу много наборов без дополнительных попарных или множественных выравниваний. В тоже время для выборки 2 число попарных совпадений пептидов тем меньше, чем больше пробы отличаются по значению pH, в 2/3 случаев это несколько десятков пептидов (рис. 1), распределённых неравномерно, и без использования набора «эталонных» пептидов получить адекватное выравнивание всего набора не получится.
Программа выравнивания работает по следующему алгоритму.
В результате программа формирует набор файлов (для каждой пробы), в которых значения RT (а также RTmin и RTmax, если они указаны) заменены соответственно на HI (HImin и HImax). Если в файле присутствовали строки, описывающие ионы, для которых не были идентифицированы пептиды, то для них также проводится перерасчёт значений RT в HI. Остальные данные переносятся из исходного файла без изменений. Объединение выровненных PF всех проб набора данных в единый файл При объединении данных результаты идентификации пептидов фиксируются в выходном файле, но не используются для самой процедуры объединения. В первую очередь программа фиксирует значение m/z иона и отбирает все ионы, отличающиеся не более чем на 0.5 ppm (параметр может варьироваться пользователем). Затем проверяется спопарное совпадение диапазонов HImin - HImax. Диапазоны считаются совпадающими, если один из них полностью перекрывает второй, либо пересечение составляет не менее 50%, а отклонение величины HI (соответствующей максимуму пика) не превышает 0.1. В выходной файл для каждого иона сохраняются параметры самого иона (m/z, заряд, HI, HImin, HImax, все величины кроме заряда усредняются), для каждой пробы формируется отдельная колонка с величиной, указанной пользователем (Area (или abundance) в нашем случае). Для каждой пробы фиксируется, был ли идентифицирован пептид в данной пробе. Если да, то приводится номер пептида из списка, также записываемого в данной строке. Если идентификация неоднозначная, то первым сохраняется вариант пептида, встречающийся чаще. Нормализация данных по величине abundance В настоящее время процедура нормализации реализована только для случая, соответствующего набору данных 1, когда для существенной части белков не должно меняться их количество от пробы к пробе. В таком случае каждый конкретный вариант иона для конкретного пептида не должен менять величину интенсивности при измерении кроме как в зависимости от состояния или калибровки аппаратной части. Зная усреднённые значения колебания интенсивности (или abundance, так как эти величины хорошо коррелируют) между пробами, можно нормировать данные для всех проб. Существует два способа определения выборки данных, по которой можно рассчитать коэффициенты, корректирующие интенсивность сигнала для разных проб. Самый простой – установить список белков, количество которых не меняется в зависимости от изменения условий, данных в эксперименте. Второй вариант – определить группу данных изменяющих значения интенсивности (или abundance) сходным образом. Группа, включающая в себя большую часть пептидов, очевидно, характеризует именно группу консервативных белков. Так как белки представлены в пробах в различном (и неизвестным заранее) количестве, то для того чтобы построить кластеры ионов пептидов со схожим распределением по пробам, требуется провести нормирование данных для каждого наблюдаемого иона. Для этого использовали следующую формулу: Fnorm = (Fi – Fmed)/(Fmed), где Fnorm – нормализованное значение величины интенсивности (или abundance) для иона пептида из i-той пробы; Fi – актуальное значение той же величины для i-той пробы; Fmed – среднее значение той же величины по всем (в данном случае 24) пробам. В данной работе для отбора кластера наибольшего размера и вычисления множителей для нормализации использовали только ионы пептидов заряда 2+, идентифицированные во всех 24 пробах набора 1 (рис. 3). Всего таких ионов было 4200. В качестве меры сходства использовали значение попарного квадрата коэффициента детерминации, пороговое значения установили равным 0.75. Всего в отобранный кластер ионов вошло 2400 значений.
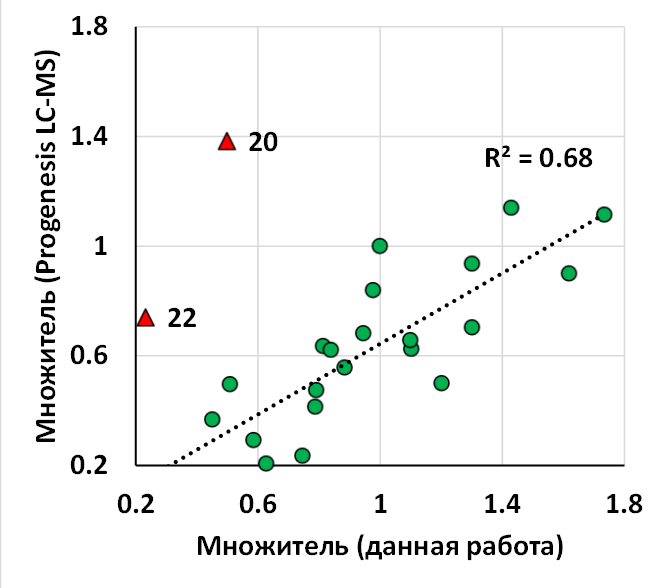
В работе [17] для набора 1 была проведена нормализация величины abundance с использованием программы Progenesis LC-MS [18]. На рисунке 4 приведено сравнение множителей для нормализации, полученных в данной работе и в работе [17]. Видно, что пробы 20 и 22 выпадают из общего тренда, однако, множители для нормализации остальных проб хорошо коррелируют между собой (R2 = 0.68).
Таким образом, представленная программа позволяет выравнивать по RT любые наборы масс-спектрометрических данных, в независимости от того, насколько они похожи по пептидному составу и количеству вещества. При этом можно отследить наличие специфических ионов и оценить их относительное количество в различных пробах. В части случаев программа позволяет провести нормализацию величины abundance, что даёт возможность делать более точные оценки при количественном анализе. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Данная работа не содержит каких-либо исследований с использованием людей и животных в качестве объектов исследования. ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021 - 2030 годы) (№ 122030100170-5). КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|