TEMPERATURE DEPENDENCE OF COLLATERAL ACTIVITY OF THERMOSTABLE RECOMBINANT CRISPR NUCLEASES Cas12b OBTAINED BY ONE-STEP PURIFICATION WITH METAL-CHELATE CHROMATOGRAPHY AFTER HETEROLOGOUS EXPRESSION
1Institute of Biomedical Chemistry, 10 Pogodinskaya str., Moscow, 119121 Russia; *e-mail: kurbatovl@mail.ru
2Chemistry Faculty of M.V. Lomonosov Moscow State University, Lenin Hills, 1/3, Moscow, 119991 Russia
Keywords:recombinant CRISPR nuclease Cas12b; single-stage purification; collateral nuclease activity; temperature dependence
DOI:10.18097/BMCRM00284
Thermostable CRISPR/Cas nucleases are considered as promising enzymes for development of next-generation DNA diagnostics by coupling loop-mediated isothermal amplification of nucleic acids (LAMP) with selective CRISPR/Cas detection of specific amplicons. In this paper, we present the results of testing the collateral activity of CRISPR nuclease AapCas12b and three variants of CRISPR nuclease BrCas12b (wild type and two mutants) obtained using simplified purification in the typical temperature range of LAMP - from 56°C to 72°C. It was shown that the use of one-step metal-chelate chromatography by excluding a stage of enzymatic removal of N-terminal sequences translated together with the target protein allows for obtaining recombinant CRISPR nucleases BrCas12b with a sufficiently high level of collateral activity. Temperature dependences of collateral activity differed among the studied BrCas12b variants. The obtained results can be useful in selecting thermostable CRISPR nucleases Cas12b for development of test systems based on a combination of LAMP and CRISPR/Cas detection.
|
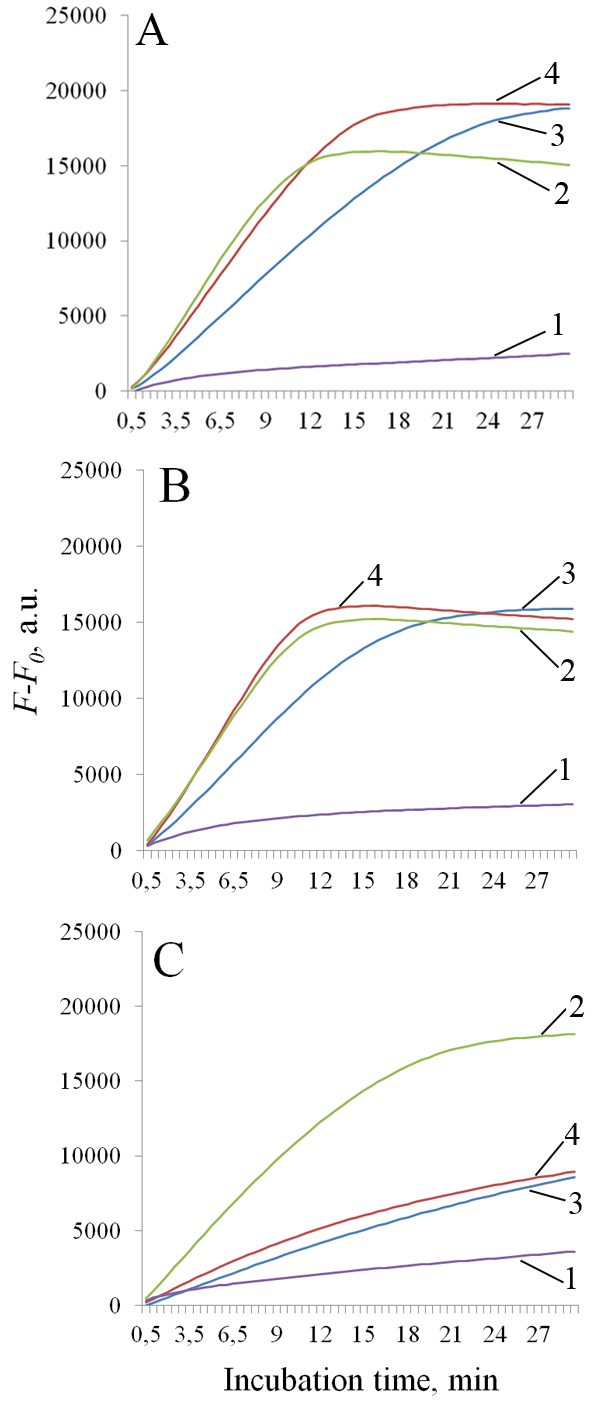
Figure 1.
Representative dependences of fluorescence on reaction time during cleavage of DNA reporters by recombinant nucleases fp-AapCas12b, fp-BrCas12b, fp-BrCas12b-FNLDTA, and fp-BrCas12b-RFND. Reaction temperature: 56°C (A), 64°C (B), 72°C (C). Curves 1, 2, 3, and 4 are fp-AapCas12b, fp-BrCas12b, fp-BrCas12b-FNLDTA, and fp-BrCas12b-RFND, respectively. The fluorescence in samples without the addition of the target DNA (F0) was subtracted from the fluorescence measured in samples where the target was present (F).
|
|
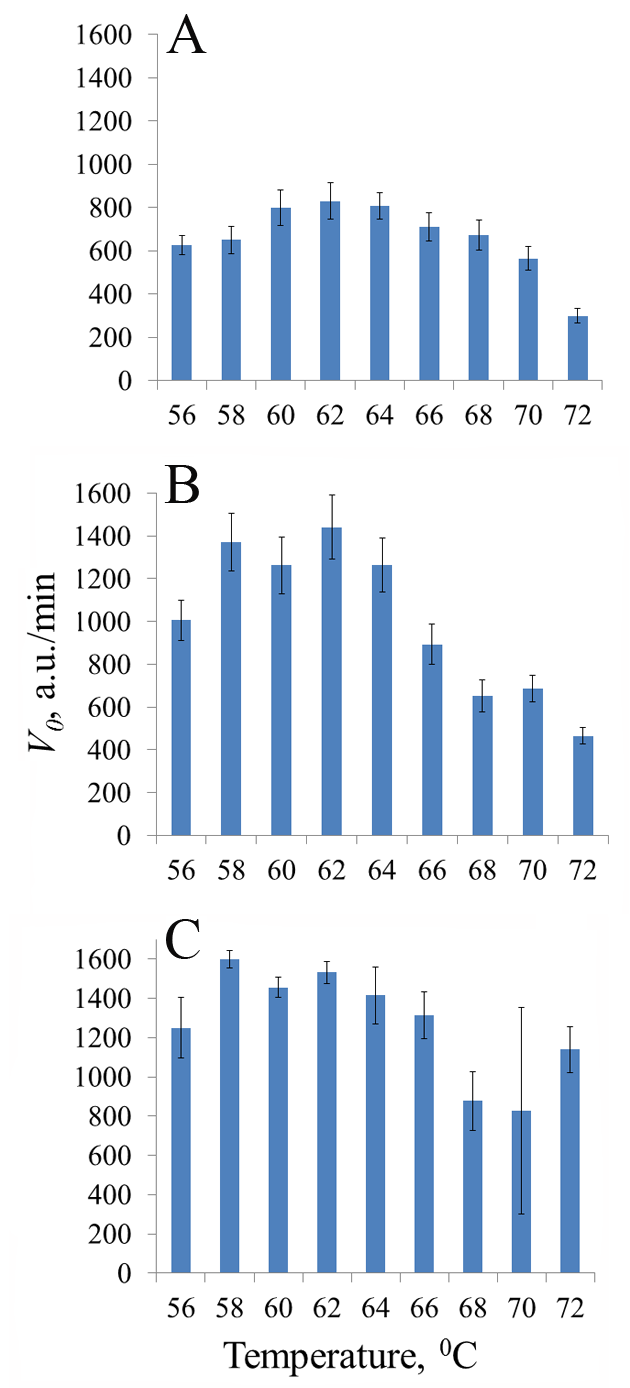
Figure 2.
Initial rate V0 of cleavage of DNA reporters by recombinant nucleases fp-BrCas12b-FNLDTA (A), BrCas12b-RFND (B), and fp-BrCas12b (C) at different reaction temperatures. The arithmetic means and standard deviations for three independent measurements are shown.
|
|
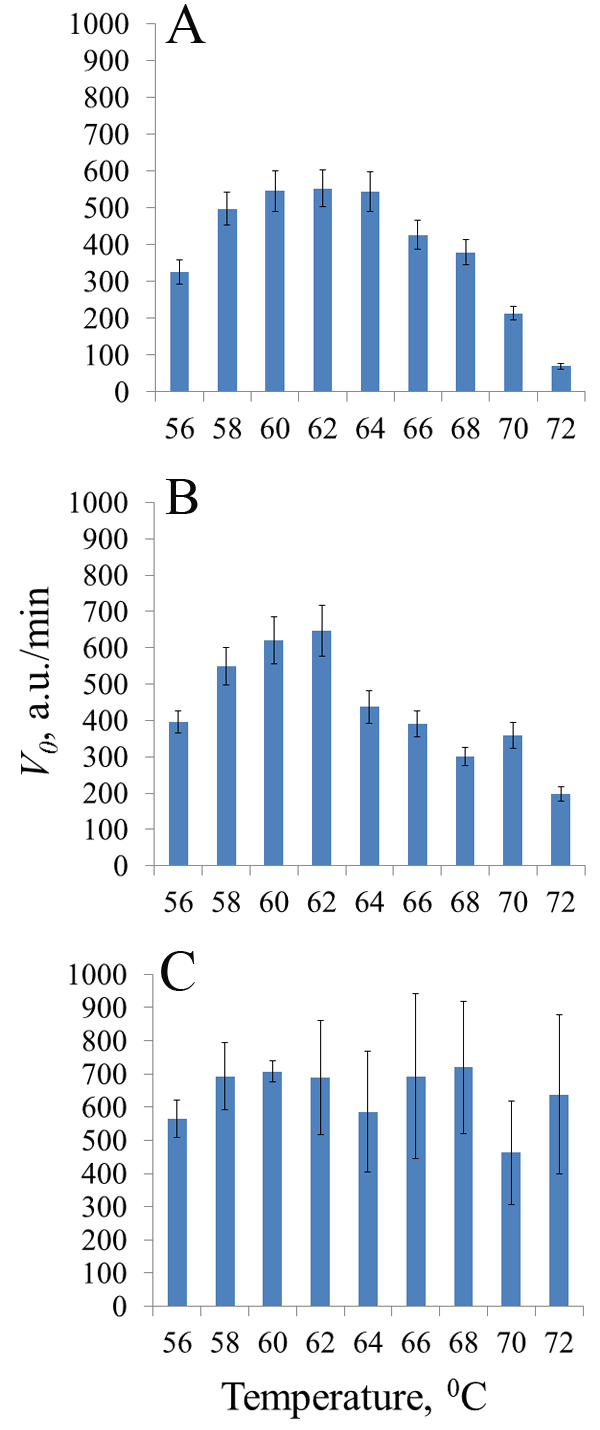
Figure 3.
Initial rate V0 of cleavage of DNA reporters by recombinant nucleases fp-BrCas12b-FNLDTA (A), BrCas12b-RFND (B) and fp-BrCas12b (C) at different reaction temperatures. Purification of the target proteins was carried out using buffer solutions from [15]. The arithmetic means and standard deviations for three independent measurements are shown.
|
Table 1. Sequences of DNA oligonucleotides used in the work. The sequence complementary to the sequence of the T7 promoter is shown in italics. The protospacer sequences of the synthetic target DNA and sequences of DNA templates, encoding spacer sequences in the guide RNA (gRNA), are highlighted in bold.
|
Name |
Sequence (5’ → 3’) |
|
T7F |
TAATACGACTCACTATAGGG |
|
gRNA |
CAGCGCTTCAGCGTTCTTCGGTGCTAACCATCTACATTTCCAC TAACTTTGGTTAGTGGAAATATAGATAGCCGTTGTGACTGAGTGACGTGTTAGGTCACC GTAGCACATGACACAACTGCACTGGTCAGCCTGTAGCTAACCACCTTCCCCTATAGTGAGTCGTATTA |
|
SARS-T-F |
TGCACAATTTGCCCCCAGCGCTTCAGCGTTCTTCGGAATGTCGCGCATTG |
|
SARS-T-R |
CAATGCGCGACATTCCGAAGAACGCTGAAGCGCTGGGGGCAAATTGTGCA |
FUNDING
The study was performed within the framework of the Program for Basic Research in the Russian Federation for a long-term period (2021–2030) (No. 122030100170-5).
REFERENCES
- van der Oost, J., Westra, E.R., Jackson, R.N., Wiedenheft, B. (2014)Unravelling the structural and mechanistic basis of CRISPR-Cas systems.Nature Review Microbiology, 12(7), 479-492. DOI
- Kaminski, M.M., Abudayyeh, O.O., Gootenberg, J.S., Zhang, F., Collins, J.J.(2021) CRISPR-based diagnostics. Nature Biomedical Engineering, 5(7), 643-656. DOI
- Fapohunda, F.O., Qiao, S., Pan, Y., Wang, H., Liu, Y., Chen, Q., Lu, P.(2022) CRISPR Cas system: A strategic approach in detection of nucleic acids.Microbiological Research, 259, 127000. DOI
- Gootenberg, J.S., Abudayyeh, O.O., Lee, J.W., Essletzbichler, P., Dy, A.J.,Joung, J., Verdine, V., Donghia, N., Daringer, N.M., Freije, C.A., Myhrvold, C.,Bhattacharyya, R.P., Livny, J., Regev, A., Koonin, E.V., Hung, D.T., Sabeti, P.C.,Collins, J.J., Zhang, F. (2017) Nucleic acid detection with CRISPR-Cas13a/C2c2. Science, 356(6336), 438-442. DOI
- Chen, J.S., Ma, E., Harrington, L.B., Da Costa, M., Tian, X., Palefsky, J.M.,Doudna, J.A. (2018) CRISPR-Cas12a target binding unleashes indiscriminatesingle-stranded DNase activity. Science, 360(6387), 436-439. DOI
- Piepenburg, O., Williams, C.H., Stemple, D.L., Armes, N.A. (2006) DNAdetection using recombination proteins. PLoS Biology, 4(7), e204. DOI
- Khmeleva, S. A., Ptitsyn, K. G., Kurbatov, L. K., Timoshenko, O. S.,Suprun, E. V., Radko, S. P., Lisitsa, A. V. (2024) Biosensing platforms forDNA diagnostics based on CRISPR/Cas nucleases: towards the detectionof nucleic acids at the level of single molecules in non-laboratory settings.Biomeditsinskaya Khimiya, 70(5), 287-303. DOI
- Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanab,e K.,Amino, N., Hase, T. (2000) Loop-mediated isothermal amplification of DNA.Nucleic Acids Research, 28(12), e63. DOI
- Joung, J., Ladha, A., Saito, M., Kim, N.G., Woolley, A.E., Segel, M.,Barretto, R.P.J., Ranu, A., Macrae, R.K., Faure, G., Ioannidi, E.I., Krajeski,R.N., Bruneau, R., Huang, M.W., Yu, X.G., Li, J.Z., Walker, B.D., Hung, D.T.,Greninger, A.L., Jerome, K.R., Gootenberg, J.S., Abudayyeh, O.O., Zhang, F.(2020) Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. The NewEngland Journal of Medicine, 383(15), 1492-1494. DOI
- Teng, F., Cui, T., Feng, G., Guo, L., Xu, K., Gao, Q., Li, T., Li, J., Zhou,Q., Li, W. (2018) Repurposing CRISPR-Cas12b for maмМalian genomeengineering. Cell Discovery, 27(4), 63. DOI
- Tian, Y., Liu, R.R., Xian, W.D., Xiong, M., Xiao, M., Li, W.J. (2020) Anovel thermal Cas12b from a hot spring bacterium with high target mismatchtolerance and robust DNA cleavage efficiency. International Journal ofBiological Macromolecules, 147, 376-384. DOI
- Nan, X., Hardinge, P., Hoehn, S., Dighe, S.N., Ukeri, J., Pease, D.F., Griffin,J., Warrington, J.I., Saud, Z., Hottinger, E., Webster, G., Jones, D., Kille, P.,Weightman, A., Stanton, R., Castell, O.K., Murray, J.A.H., Jurkowski, T.P.(2023) VarLOCK: sequencing-independent, rapid detection of SARS-CoV-2variants of concern for point-of-care testing, qPCR pipelines and nationalwastewater surveillance. Scientific Reports, 13(1), 20832. DOI
- Nguyen, L.T., Macaluso, N.C., Pizzano, B.L.M., Cash, M.N., Spacek, J.,Karasek, J., Miller, M.R., Lednicky, J.A., Dinglasan, R.R., Salemi, M., Jain,P.K. (2022) A thermostable Cas12b from Brevibacillus leverages one-potdiscrimination of SARS-CoV-2 variants of concern. EBioMedicine, 77, 103926. DOI
- Nguyen, L.T., Rananaware, S.R., Yang, L.G., Macaluso, N.C., Ocana-Ortiz,J.E., Meister, K.S., Pizzano, B.L.M., Sandoval, L.S.W., Hautamaki, R.C., Fang,Z.R., Joseph, S.M., Shoemaker, G.M., Carman, D.R., Chang, L., Rakestraw,N.R., Zachary, J.F., Guerra, S., Perez, A., Jain, P.K. (2023) Engineering highlythermostable Cas12b via de novo structural analyses for one-pot detection ofnucleic acids. Cell Reports Medicine, 4(5), 101037. DOI
- Kurbatov, L.K., Radko, S.P., Kravchenko, S.V., Kiseleva, O. I., Durmanov, N.D., Lisitsa, A. V. (2020) Single Stage Purification of CRISPR/Cas13a Nucleasevia Metal-Chelating Chromatography Following Heterologous Expression withthe Preservation of Collateral Ribonuclease Activity. Applied Biochemistry andMicrobiology, 56(6), 671–677. DOI
- Kurbatov, L. K., Radko, S. P., Khmeleva, S. A., Ptitsyn, K. G., Timoshenko,O. S., Lisitsa, A. V. (2024) Application of DETECTR for Selective Detection ofBacterial Phytopathogen Dickeya solani Using Recombinant CRISPR-NucleaseCas12a Obtained by Single-Stage Chromatographic Purification. AppliedBiochemistry and Microbiology, 60(1), 17-25. DOI
- Habimana, J.D., Mukama, O., Chen, G., Chen, M., Amissah, O.B., Wang,L., Liu, Y., Sun, Y., Li, A.L., Deng, S., Huang, J., Yan, X.X., Rutaganda, T.,Mutangana, D., Wu, L.P., Huang, R., Li, Z. (2023) Harnessing enhancedCRISPR/Cas12a trans-cleavage activity with extended reporters and reductantsfor early diagnosis of Helicobacter pylori, the causative agent of peptic ulcersand stomach cancer. Biosensors and Bioelectronics, 222, 114939. DOI
- Ptitsyn, K. G, Khmeleva, S. A, Kurbatov, L. K, Timoshenko, O. S, Suprun,E. A, Radko, S. P, Lisitsa, A. V. (2024). Lamp Primer Designing Software: TheOverview. Biomedical Chemistry: Research and Methods, 7(4), e00226. DOI
- Kurbatov, L. K., Radko, S. P, Khmeleva, S. A., Timoshenko, O. S. Lisitsa,A. V. (2022) Standardization of Recombinant CRISPR/Cas13a-nucleasePreparations by Using RNase A of Known Activity. Biomedical Chemistry:Research and Methods, 5(4), e00177. DOI
- Hand, T.H., Das, A., Li, H. (2019). Directed evolution studies of athermophilic Type II-C Cas9. Methods in Enzymology, 616, 265–288. DOI
- Kumar, S., Tsai, C.J., Nussinov, R. (2000) Factors enhancing proteinthermostability. Protein Engineering, 13(3), 179–191. DOI