|
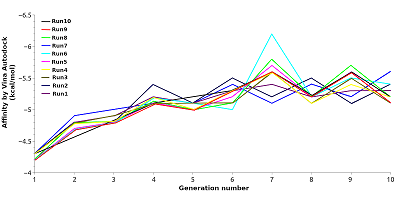
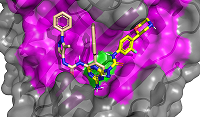
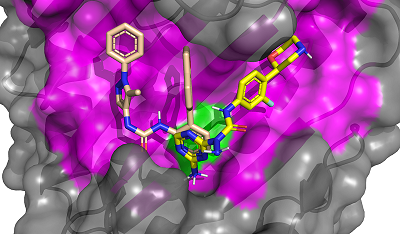
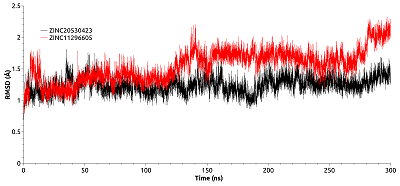
CONTENTS COMPLIANCE WITH ETHICAL STANDARDS Figure 1 Average values of the predicted affinities for the generated ligands. Figure 2 Compounds 1 and 2 in the groove on the surface of HSP90 near the Thr-90 residue. Figure 3 Compounds used for design pharmacophore models. Figure 5 . RMSDs of compounds 19 (ZINC20530423) and 21 (ZINC1126605) during molecular dynamics. Table 1Initial pharmacophore models. Table 2Secondary pharmacophore models used for pharmacophore search. Table 3Calculated binding free energies using MM–PBSA for complex HSP90 with compounds 19 and 21. ypes of chemical reactions in xenobiotic metabolism (adapted from [6]) Table 4Oral bioavailability (Lipinski’s rules) of selected compounds. |
A Way for Finding Ligands for New Binding Sites 1Institute of Biomedical Chemistry, Pogodinskaya Street, 10, Moscow 119121, Russia; e-mail: veselov@ibmh.msk.su 2Pirogov Russian National Research Medical University, Moscow, Russia Key words: molecular docking, binding site, pharmacophore model, molecular dynamics, HSP90, design de novo DOI: 10.18097/BMCRM00200 INTRODUCTION
Computer-aided drug design (CADD) is one of the main approaches to the discovery and optimization of new ligands with the desired biological activity. A rather complicated situation may arise when ligands that bind the desired region of the protein are unknown. At present, numerous methods have been proposed to identify the potential druggable binding sites. The prediction of such sites can be the first step in drug discovery projects [1-5]. Such situation often occurs when interface of a protein–protein complex [6] or predicted allosteric sites [7] are used as targets. The main approach currently used to find ligands and to estimate their affinity for new binding sites, when known ligands are absent, is molecular docking. However, the docking scoring functions have a poor ability to correctly predict affinity of ligands [8]. It results in numerous false positive and false negatives docking results [9-11]. One of the possible ways to overcome this problem is to identify structural fragments that are able to interact with the desired regions of the binding site. A similar approach is used in a fragment-aided drug design method. The obtained information about such fragments can be used to select compounds from molecular databases during the virtual screening. Prostate cancer (PC) is one of the main causes of death among men in developed countries [12]. The numerous efforts were aimed to identify the mechanisms of pathogenesis and to develop methods of prevention and treatment of this disease. At present, there are two main therapeutic approaches to treat PC. They include inhibitors of CYP17A1 (abiraterone) and antagonists of the androgen receptors (AR) (cyproterone, flutamide, bicalutamide) [13, 14]. In the cytoplasm, AR is complexed with the chaperone HSP90 and other proteins. Binding of an agonist to the AR leads to conformational changes in the protein followed by dissociation of the AR-chaperone complex and subsequent receptor translocation into the nucleus. Recently it has been shown that dissociation of the AR-HSP90 complex requires additional phosphorylation of Thr-90 residue of HSP90 [15]. Phosphorylation is carried out by the protein kinase PKA 1. Therefore, the design of compounds that prevent Thr-90 phosphorylation of HSP90 may become a new promising direction in PC therapy. Here, we describe an approach to search for ligands to a new binding site for which ligands are unknown. This approach includes generation of compounds at the selected binding site by the de novo design method, followed by generation of pharmacophore models based on designed compounds and pharmacophore-based screening of a database. At the next stage, the compounds that fit the pharmacophore model are docked to this binding site. The complexes, which were top-ranked by docking position and scoring function, were then proceeded for molecular dynamics simulations to estimate their stability. This approach has been tested for searching compounds that may prevent HSP90 phosphorylation. MATERIALS AND METHODS The spatial structure of HSP90 (PDB code 3T0H, resolution 1.2 Å) was downloaded from PDB [16]. De novo design of compounds was done by AutoGrow4 program [17]. AutoGrow4 starts from the initial population of compounds. This initial population, defined as "generation 0", consists of chemically diverse molecular fragments from the AutoGrow library. During the next step, the first generation of compounds are designed by applying three operators to the structural fragments of the "generation 0" population: elitism, mutation, and crossover. Subsequent generations of compounds are designed similarly to the previously generation. The elitism operator transfers subpopulations of the fittest compounds from previous generation to the next generation without change. The mutation operator performs a virtual chemical reaction, which modifies the parent compound to a new one. Seventy nine of the 94 standard AutoGrow4 reactions use two fragments. One of the reagents is taken from the previous generation, and the other one is taken from the AutoGrow4 molecular fragment libraries. The crossover operator combines two fragments taken from the previous generation into one new compound. To evaluate the affinity of molecular fragments and designed compounds, molecular docking is used. By default, the docking program is QVina, and the scoring function is the standard scoring function of Vina AutoDock. Pharmacophore models were generated using the PharmaGist web service [18]. The following values of weights of pharmacophore features were used for the pharamcophore design: aromatic - 5.0; anion/cation - 3.0; H-bond donor/acceptor - 3.0 and hydrophobic – 1.0 (the maximum value for each weight is 10.0). The ZINCPharmer web service was used to search for potential ligands in the ZINC12 database [19]. Molecular docking was done using AutoDock Vina [20]. AutoDock Tools package was used to set docking parameters. Binding poses were selected based on their binding affinities calculated by the Autodock Vina scoring function. Analysis of binding poses was perform using the PLIP server [21]. Molecular dynamics calculations (MD) were performed using the Gromacs2020 software package [22]. The explicit solvent (TIP3P) with addition of Na+ and Cl- ions was used to neutralize the system. The Amber99-ILDN forcefield [23] was used for atomic parametrization of protein molecules and GAFF forcefield for ligands. The systems of solvated protein-ligand complexes were minimized for 50 000 steps. All systems were equillibrated in NVT- and NPT ensembles for 5 ns. Productive dynamics were simulated on trajectories of 300 ns with a step of 2 fs using the Berendsen thermostat [24] and the Parrinello–Rahman barostat [25] for control of pressure (1 atm) and a temperature of the system (300 K). Long-range electrostatic interactions were treated with the PME method. The cut-off value for short-range non-bonded interactions was set to 12Å. The LINCS algorithm was used to constraint the bonds with hydrogen. Trajectory analysis was performed using the Gromacs-2020 built-in tools and VMD-1.9.1 software. The molecular mechanics combined with the generalized Born and surface area continuum solvation (MM/GBSA) method was used to calculate free energies of ligand-protein complexes. The main idea of the MM/GBSA method is to calculate separately the ligand solvation free energy, the receptor solvation free energy, the complex solvation free energy, and the interaction energy of the receptor and ligand in vacuum. The free energy of interaction between the ligand and the receptor ΔGbind can be calculated using equation 1: ΔGbind = ΔH-TΔS= (ΔEMM+ΔGsolv)-T*ΔS (1), where ΔGsolv = ΔGp + ΔGnp (2) ΔEMM = ΔEele + ΔEvdW (3) ΔEMM is the interaction energy between the ligand and the receptor, calculated on the basis of molecular mechanics, ΔEele and ΔEvdW are the contributions of electrostatic and van der Waal energy, respectively. ΔGsolv is the solvation free energy; ΔGp and ΔGnp are the contributions of polar and nonpolar interactions to the solvation, respectively. -T*ΔS is the change in conformational entropy upon binding, -T*ΔS is not considered in the current work because of a high computational cost of entropy calculations and the inclusion of this term does not always improve the accuracy of calculations. To calculate the enthalpy contribution (ΔH= ΔEMM+ΔGsolv) 500 frames of the last 50 ns of dynamics were used. RESULTS AND DISCUSSION In this study, we have tested an applicability of the approach to search ligands to the new binding sites for which ligands are unknown. This approach is based on a pilot generation of new compounds by a program for a de novo ligand design. During the next step, a set of generated compounds is used to design a pharmacophore model. The obtained pharmacophore model is further used to search potential ligands in a molecular database. The resulting focused library of compounds is docked to the potential binding site. Such procedure for selection of the focused library makes it possible to avoid docking of large databases and thus can significantly reduce time costs. The AutoGrow4 was used to design ligands based on the three-dimensional structure of the binding site of HSP90, located near Thr-90 (phosphorylation of this residue facilitates dissociation of the AR complex with HSP90). The standard AutoGrow4 library consisting of ~6100 molecular fragments (weight 100-150 Da) was used as the initial population for the “generation 0”. During the first generation, the operators “mutation” and “crossover” were applied 500 times, in the second and subsequent generations these operators were used 2500 times. The elitism operator selected 500 compounds from each previous generation to transfer them to the next one. There were ten independent runs of Autogrow4, each of which consisted of ten generations. During each generation, more than forty thousand compounds were generated. The mean and median values of the affinities of the generated compounds increased up to the sixth generation in the each run; they then fluctuated around -5.4 kcal/mol for the average and -5.3 kcal/mol for the median affinities (Figures 1 and 1S, Supplementary materials, respectively). The maximum affinity values increased until the fourth or the fifth generation, and then the values reached a plateau (Figure 2S, Supplementary materials). Thus, we conclude that ten generations are enough to generate compounds with the maximum predicted affinity for the protein of interest.
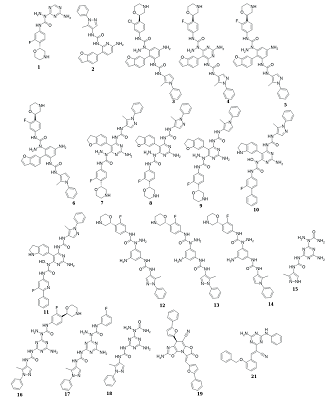
However, the generated compounds are absent in available molecular databases. To avoid the synthesis of compounds in the early stages of a discovery project, the similar compounds may be found in molecular databases based on pharmacophore models. The compounds with the highest predicted affinities in each generations were selected for analysis. Visual inspection of their poses in the binding site showed that most of the generated compounds (~30%) had two similar fragments: an aromatic or cyclic aliphatic fragment with a nitrogen atom attached to it directly or via one carbon atom. This nitrogen atom was located near Thr-90 and formed a hydrogen bond with its hydroxyl group . Two molecules generated by AutoGrow (1 and 2) located along the groove with nitrogen atoms oriented towards Thr-90 were considered as the basis for generating the pharmacophore (Fig. 2). Several compounds (3 – 18) were also selected because of the aforementioned substructure and similar position in the binding site (Fig. 3). These compounds were additionally docked to the binding site to obtain alignment and conformations required to design pharmacophore models. The poses of the docked molecules coincided with the position of the compounds 1 and 2.
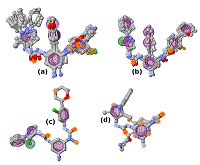
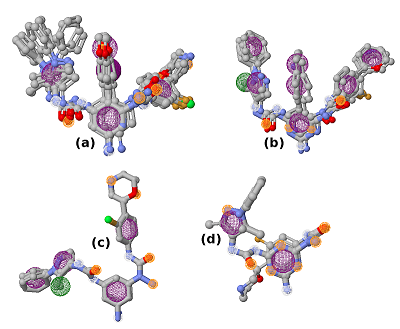
Compounds 1 – 18 were used to generate pharmacophores using the PharmaGist web service. Initially, four pharmacophore models were designed (Table 1). Pharmacophore 1 (Fig. 4a) was generated on the basis of molecules 1-6, pharmacophore 2 - molecules 7 - 11 (Fig. 4b), pharmacophore3 - molecules 12 - 14 (Fig. 4c) and pharmacophore 4 - molecules 15 – 18 (Fig. 4d). However, the obtained pharmacophore models were too complex, and no one compound satisfying these pharmacophore models was found in database. This prompted us to simplify these models for the second generation of pharmacophore models that contained 5-7 pharmacophore features (Table 2). The resultant seven secondary pharmacophore models (Figure 3S, Supplementary materials) were used for pharmacophore searches in the ZINC database using the ZINCPharmer web service. These searches yield:
Thus, 794 compounds fitted the pharmacophore models were selected.
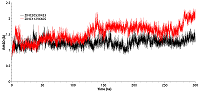
Molecular docking was used for initial estimation of the ability of selected compounds to interact with binding site. The criteria for selecting the docked molecules for subsequent analysis based on the following: the values of the scoring function were less than -6.0 kcal/mol; molecules formed H-bonds with the Thr-90; and the position of the molecules in the binding site (preference was given to ones that occupied >50% of the cavity surface area). Sixteen compounds were selected. To determine the stability of the obtained complexes, simulations of molecular dynamics were carried out. Most compounds flowed into solution after 2–3 ns simulation, and only two complexes (with compounds 19 and 21) were stable. Compounds 19 and 21 have a common structural element described above: an aromatic ring with an amino group attached to it. The amino groups of both compounds were located in a cavity near Thr-90 and formed the H-bond with it. In the first 10 ns of simulation, the RMSD values of the compounds 19 and 21 reached 9Å. This indicates a shift of the compounds during the dynamics from the initial positions predicted by docking (Fig. 5).
The last 50 ns of the trajectory was used to analyze the H-bonds formed between the protein and compounds 19 and 21. Compound 21 formed H-bonds with the residues Thr-90, Asn-83, Val-222, Glu-223, Glu-178, Lys-224. Although compound 21 retains the H-bond with the Thr-90 side chain, this bond had low stability (7.20% occupancy). Compound 21 formed the most stable H-bond with the oxygen atom of the main chain Val-222 (occupancy 33.91%). Compound 19 formed H-bonds with Asn-83, Glu-223, and Lys-224 residues. The most stable H-bonds was between the amino group of compound 19 and Glu223 side chain (occupancy 30.38%). Thus, it appears that the Thr-90 residue plays an insignificant role in the binding of the studied ligands. Additionally, to evaluate the affinity of compounds 19 and 21 for HSP90, the binding free energies of HSP90-19 and HSP90-21 complexes were calculated using the MM-GBSA method. The binding energies for compound 19 and 21 were -26.35 kcal/mol and -26.97 kcal/mol, respectively (Table. 3). To identify the residues that make the greatest contribution to binding, the per-residue free energy decomposition was performed. The residues which contribution ≤-1 kcal/mol were taken into the consideration. For compound 19 these residues included Asn-79, Ile-81, Asn-83, Val-92, and Phe-221, and for the compound 21 these residues included Ile-81, Thr-90, Val-92, and Phe-221. For the both complexes, Ile-81 made the greatest contribution to the interaction energy. Polar Asn-79 and Asn-83 make a significant contribution to the binding of the compound 19. For the compound 21 the only polar residue whose interaction energy with the ligand ≤-1 kcal/mol was Thr90. Calculation of ADMET properties showed that both compounds satisfied the Lipinski's rules (Table 4).
The proposed approach allowed us to identify two compounds that are potentially able to bind to HSP-90 near the Thr-90 residue and to prevent its phosphorylation required for the activation of androgen receptor. Method of de novo ligand design made it possible to determine the key functional groups required for binding to the considered region of the protein. Based on these functional groups we designed several pharmacophore models which allowed to identify 794 compounds from ZINC12 database, that fitted to the pharmacophores. These compounds were used for further docking to evaluate their binding affinities. It can be seen that our approach allowed to reduce the total number of docked compounds from hundreds of thousands or even millions molecules to less than 800 structures. It is also worth saying that MD simulations are the crucial step to evaluate the stability and binding affinity of obtained complexes. Thus, out of 16 complexes selected during the docking, only two of them turned out to be stable in the course of molecular dynamics. The developed approach can be used for discovery of compounds for other proteins, for which information of their ligands is absent. CONCLUSIONS The protein structures may contain a number of potential ligand binding sites (protein-protein interfaces, allosteric sites) that may participate in modulation of target protein functions and be potential sites for drug development. The lack of known ligands and favourable substructures complicates the application of computational approaches. In the case of virtual screening based on molecular docking, the effective selection of potential ligands based on scoring functions is limited by their accuracy. Here, we have considered the approach aimed at solving such problems. It is based on the method of de novo ligand design. It can be expected that repeated de novo design of ligands started from random substructures will reveal favorable fragments of ligands and their positions in the binding site. The designed virtual molecules containing these favourable fragments allow to design pharmacophore models that can be used for preliminary selection of molecules in molecular databases. The resultant set of molecules docked in the binding site to check the ability to accommodate in the binding site. The additional criterion for correct binding pose is the coincidence of the identified favorable fragments in the same region of the binding site that were obtained during de novo design. Subsequent evaluation of the selected ligands can be carried out by standard CADD methods, including molecular dynamics, estimation of binding energy, etc. COMPLIANCE WITH ETHICAL STANDARDS This article does not contain any research involving humans or using animals as objects. FUNDING This work was supported by Russian Foundation for Basic Research (Grant number 19-34-90057). CONFLICT OF INTEREST The authors declare no conflict of interest Supplementary materials are available at http://dx.doi.org/10.18097/BMCRM00200 REFERENCES
|