A Program for Predicting the Retention Time of Peptides with Post-Translational Modifications
Institute of Biomedical Chemistry, Pogodinskaya Street, 10, Moscow 119121, Russia; *e-mail: an.voronina@list.ru
Keywords:peptide retention time; isoelectric point; post-translational modifications; web service
DOI:10.18097/BMCRM00196
This paper describes the Retention Time Predictor (RTP) program and web service for predicting the retention time of peptides on a chromatographic column in mass spectrometry experiments. Taking into account post-translational modifications of peptides the program represents a modification of the well-known SSRCalc version 3 (Krokhin, Anal. Chem. 2006, 78(22), 7785-7795). The values of retention coefficients for modified amino acid residues and the algorithm for calculating the isoelectric point value were from the pIPredict program (Skvortsov et al., Biomed. Chem. Res. Meth. 2021, 4(4), e00161). Modifications described in the program include (i) Tandem Mass Tag (TMT) and Isobaric Tags for Relative and Absolute Quantification (iTRAQ) labels; (ii) acetylation, formylation, and methylation of the N-terminal residue and/or lysine side chain; (iii) carbamidomethylation of cysteine, asparagine, and glutamic acid residues; (iv) oxidation and double oxidation of methionine and proline residues; (v) phosphorylation of serine, threonine, and tyrosine residues; (vi) C-terminal amidation of lysine and arginine residues; (vii) formation of propionamide with a cysteine residue. Retention coefficient estimation was based on data from 25 mass spectrometry experiments for which identification was performed from the raw data deposited in the ProteomeXchange database. The RTP program and web service are freely available at http://lpcit.ibmc.msk.ru/RTP.
|
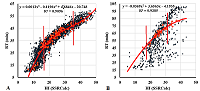
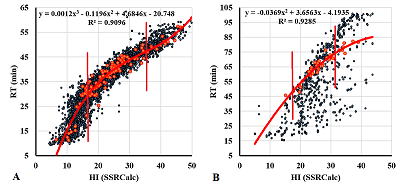
Figure 1.
Examples of correlations between the observed RT values and HI values calculated by the SSRCalc program for samples S5 (A) and S22 (B). One of the fractions for which the trend line parameters are given is highlighted in red. Vertical lines limit the area of linear dependence.
|
|
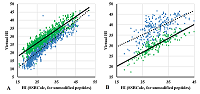
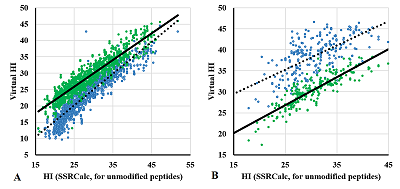
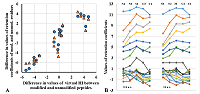
Figure 2.
Comparison of the virtual Hi value for peptides of the same sequence with PTM (blue) and without (green) with the Hi value calculated by SSRCalc for peptides without modifications.
A. M(+15.99) modification at the M position; B. N-terminal modification [+42.01]. |
|
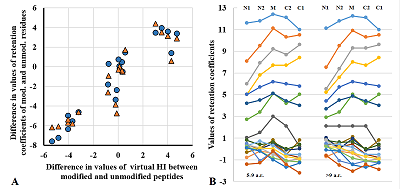
Figure 3.
A. Comparison of the averaged values of the bias of the virtual HI for peptides with the same amino acid sequence after PTM addition with the value of the retention coefficient determined in this work. The values calculated for sets of peptides with a single PTM are shown in blue, and those calculated for peptides with more than one PTM are shown in orange. B. Comparison of retention factor values for 20 canonical amino acid residues set in the SSRCalc program for different positions in the peptide.
|
|
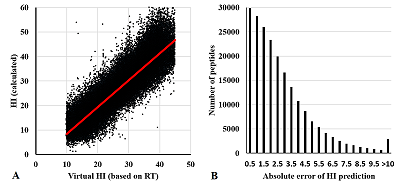
Figure 4.
Comparison of the calculated and actual HI value (A) and absolute error distribution (B) for the complete data set including peptides with more than one PTM.
|
|
CLOSE

|
Table 1.
Description of the 25 mass spectrometry experiment data sets used in this work.
|
|
CLOSE
|
Table 2.
Number of observations for peptides with different modifications for the sets to which retention factor values were fitted.
|
|
CLOSE
|
Table 3.
Retention coefficient values from this paper for use in the RTP program.
|
FUNDING
The work was performed within the framework of the Program for Basic Research in the Russian Federation for a long-term period (2021-2030) (№122030100170-5).
REFERENCES
- Bączek, T., Kaliszan, R. (2009) Predictions of peptides' retention times in reversed‐phase liquid chromatography as a new supportive tool to improve protein identification in proteomics, Proteomics, 9(4), 835-847. DOI
- Krokhin, O. (2012) Peptide retention prediction in reversed-phase chromatography: proteomic applications. Expert Review of Proteomics, 9(1), 1-4. DOI
- Krokhin, O.V. (2006) Sequence-specific retention calculator. Algorithm for peptide retention prediction in ion-pair RP-HPLC: application to 300-and 100-Å pore size C18 sorbents. Analytical Chemistry, 78(22), 7785-7795. DOI
- Wilburn, D.B., Shannon, A.E., Spicer, V., Richards, A.L., Yeung, D., Swaney, D.L., Krokhin, O.V., Searle, B.C. (2023) Deep learning from harmonized peptide libraries enables retention time prediction of diverse post translational modifications, bioRxiv 5(30), 542978. DOI
- Hemshekhar, M., Faiyaz, S., Choi, K. Y. G., Krokhin, O. V., Mookherjee, N. (2019) Immunomodulatory functions of the human cathelicidin LL-37 (aa 13–31)-derived peptides are associated with predicted α-helical propensity and hydrophobic index. Biomolecules, 9(9), 501. DOI
- Ma, B., Zhang, K., Hendrie, C., Liang, C., Li, M., Doherty-Kirby, A., & Lajoie, G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Communications in Mass Spectrometry: RCM, 17(20), 2337–2342. DOI
- Skvortsov, V.S., Voronina, A.I., Ivanova, Y.O., Rybina, A.V. (2021) The predic-tion of the isoelectric point value of peptides and proteins with a wide range of chemical modifications. Biomedical Chemistry: Research and Methods, 4(4), e00161. DOI
- Branca, R., Orre, L., Johansson, H., Granholm, V., Huss, M., Pérez-Bercoff, A., Forshed, J., Käll, L., Lehtiö, J. (2014) HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat Methods, 11, 59–62. DOI
- Panizza, E., Branca, R. M. M., Oliviusson, P. et al. (2017) Isoelectric point-based fractionation by HiRIEF coupled to LC-MS allows for in-depth quantitative analysis of the phosphoproteome. Scientific Reports, 7, 4513. DOI
- Zhu, Y., Orre, L. M., Johansson, H. J. et al. (2018) Discovery of coding regions in the human genome by integrated proteogenomics analysis workflow. Nat Commun, 9, 903. DOI
- Panizza, E., Zhang, L., Fontana, J. M., Hamada, K., Svensson, D., Akkuratov, E. E., Scott, L., Mikoshiba, K., Brismar, H., Lehtiö, J., & Aperia, A. (2019) Ouabain-regulated phosphoproteome reveals molecular mechanisms for Na+, K+-ATPase control of cell adhesion, proliferation, and survival. FASEB journal: official publication of the Federation of American Societies for Experimental Biology, 33(9), 10193–10206. DOI
- Babačić, H., Lehtiö, J., Pico de Coaña, Y., Pernemalm, M., & Eriksson, H. (2020) In-depth plasma proteomics reveals increase in circulating PD-1 during anti-PD-1 immunotherapy in patients with metastatic cutaneous melanoma. Journal for immunotherapy of cancer, 8(1), e000204. DOI
- Kiseleva, O, Zgoda, V, Naryzhny, S, Poverennaya, E. (2020) Empowering Shotgun Mass Spectrometry with 2DE: A HepG2 Study. International Journal of Molecular Sciences, 21(11), 3813. DOI